

 GCP(グッド・クリニカル・プラクティス)とは?
GCP(グッド・クリニカル・プラクティス)とは?

GCPとは、治験コーディネーター(CRC)が遵守すべき規則が記載された省令で、法律に準じるもの。
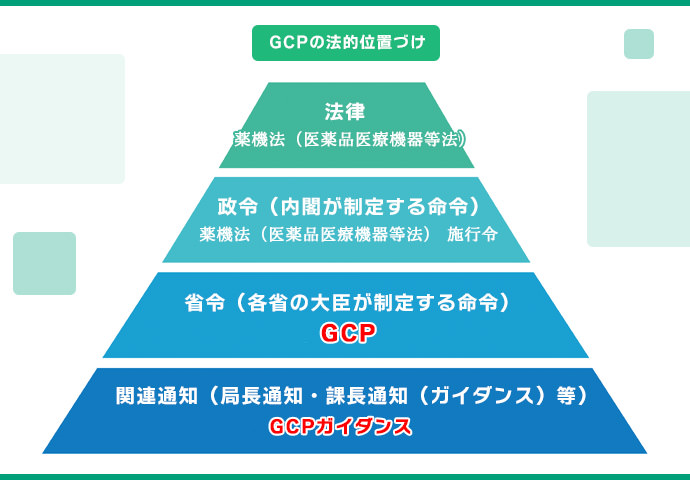
治験の進行に関する基本的なルールは、「医薬品の臨床試験の実施基準」、通称GCP(グッド・クリニカル・プラクティス)と言われます。
国内のGCPは、厚生労働省がまとめたもので、欧米諸国をはじめとする世界中で認められている医薬品開発における国際的なルールを反映しています。
治験の国際的な基準としては、医薬品規制調和国際会議(ICH)で合意されたICH-GCPがあり、世界各国でこれに準拠したGCPが整備されています。
日本のGCPもICH-GCPの原則を踏まえながら、国内の法制度や医療環境に適した形で策定されています。日本では1997年3月に初めてGCPが公表され、その後、2005年4月には医療機器GCPが、2018年6月には再生医療等製品GCPが新設されています。
GCPが制定される以前は、1989年10月2日に通知され、1990年10月に施行された旧GCPがありました。これは「通達」であり、法的拘束力はありませんでした。
しかし、1997年3月に公表されたGCPは「省令(厚生省令第28号)」として制定されました。そのため、治験を行う際にこれに違反した企業や医療機関は、治験の中止や新薬販売の中止といった法的な処分を受ける可能性があります。
その後、GCPは医薬品GCPに加えて、2005年に医療機器GCP、2014年に再生医療等製品GCPが新たに制定されました。
ICH-GCPと省令GCPの違い 旧GCPと新GCPの違い 各GCPの違い 面接を受ける前に目を通しておくべき部分 GCP(厚生労働省令第28号)の詳細
 ICH-GCPと省令GCP(日本のGCP)の主な違い
ICH-GCPと省令GCP(日本のGCP)の主な違い
ICH-GCPと省令GCP(日本のGCP)の主な違いは以下になります。
- 1.
- 日本のGCPでは、治験審査委員会(IRB)の委員は5名以上と具体的な人数を定めています。一方、ICH-GCPでは、最低委員数の具体的な規定はなく、より柔軟な運用が可能となっています。
- 2.
- 日本のGCPでは、治験責任医師は「医師または歯科医師」と明確に規定されています。一方、ICH-GCPでは、各国の法規制に従うとされ、より広い範囲の医療専門職が認められる場合があります。
- 3.
- 日本のGCPでは、治験実施医療機関の要件や手順について詳細な規定があります。一方、ICH-GCPでは、より一般的な要件が示されており、各国の状況に応じた柔軟な対応が可能となっています。
旧GCPと新GCPの主な違い
旧GCPと新GCPの主な違いは以下の6点です。
- 1.
- 旧GCPでは、被験者の同意を口頭で得ることができましたが、新GCPでは文書による同意の取得が義務づけられました。
- 2.
- 旧GCPでは治験総括医師が治験実施計画書(プロトコール)を作成していましたが、新GCPでは製薬企業が作成することになりました。
- 3.
- 製薬企業による治験のモニタリングや監査の実施が義務づけられました。
- 4.
- 新GCPでは、治験を行う各医療機関に治験責任医師を配置することが規定されました。
- 5.
- 治験審査委員会(IRB)の機能強化が図られ、その医療機関と利害関係のない人物が委員として参加することが必須とされました。
- 6.
- 旧GCPは通達でしたが、新GCPは省令となり、薬機法(旧薬事法)にも記載され、治験の法律上の位置づけが強化されました。
そして、このGCPを遵守して治験を行うために、治験コーディネーター(CRC)という新たな職種が誕生しました。
医薬品GCP、医療機器GCP、再生医療等製品GCPの主な違い
医薬品GCPと医療機器GCP、再生医療等製品GCPの主な違いは以下になります。
- 1.
- 医療機器の治験では、薬剤師や看護師に加えて、診療放射線技師、臨床検査技師、臨床工学技士なども実務に関わることが多いです。そのため、医療機器GCPでは、これらの専門職種も「治験協力者」および「医療機関で専門知識を有する職員」として含まれます。
- 2.
- 医療機器の治験では、被験者だけでなく、医師、技師、看護師などの治験関係者にも、被曝や感電などの有害な事象が起こる可能性があります。そのため、医療機器GCPでは、被験者以外の医療関係者に生じた疾病や障害も有害事象として扱います。
- 3.
- 医療機器と再生医療等製品の治験では、治験依頼者が実施医療機関に治験機器や治験製品を提供する際、必要に応じて医療機関のスタッフに対して使用方法などの教育訓練を行う必要があります。
- 4.
- 医療機器と再生医療等製品の治験は、医薬品と比べて実施件数が非常に少ないという特徴があります。そのため、治験機器や治験製品の開発およびモニタリングを担当する部門の職員でも、当該治験に直接関わっていなければ監査業務を行うことができます。
 面接を受ける前に目を通しておくべき部分
面接を受ける前に目を通しておくべき部分
現在のGCPは6章より構成され、重要なものは次の3章です。
- 第2章 治験の準備に関する基準
- 第3章 治験の管理に関する基準
- 第4章 治験を行う基準
治験コーディネーター(CRC)への転職を希望される方は、まず第4章の「治験を行う基準」に目を通しておきましょう。
 GCP(厚生労働省令第28号)の詳細
GCP(厚生労働省令第28号)の詳細
医薬品の臨床試験の実施の基準に関する省令
(平成九年厚生省令第二十八号)
薬事法(昭和三十五年法律第百四十五号)第十四条第三項(同条第六項、同法第十九条の二第四項及び第二十三条において準用する場合を含む。)、第十四条の四第四項並びに第十四条の五第四項(これらの規定を同法第十九条の四及び第二十三条において準用する場合を含む。)、第八十条の二第一項、第四項及び第五項並びに第八十二条の規定に基づき、医薬品の臨床試験の実施の基準に関する省令を次のように定める。
--------------
第一章 総則(第一条―第三条)
第二章 治験の準備に関する基準
第一節 治験の依頼をしようとする者による治験の準備に関する基準(第四条―第十五条)
第二節 自ら治験を実施しようとする者による治験の準備に関する基準(第十五条の二―第十五条の九)
第三章 治験の管理に関する基準
第一節 治験依頼者による治験の管理に関する基準(第十六条―第二十六条)
第二節 自ら治験を実施する者による治験の管理に関する基準(第二十六条の二―第二十六条の十二)
第四章 治験を行う基準(第二十七条―第五十五条)
第一節 治験審査委員会(第二十七条―第三十四条)
第二節 実施医療機関(第三十五条―第四十一条)
第三節 治験責任医師(第四十二条―第四十九条)
第四節 被験者の同意(第五十条―第五十五条)
第五章 再審査等の資料の基準(第五十六条)
第六章 治験の依頼等の基準(第五十七条―第五十九条)
--------------
第一章 総則
(趣旨)
第一条 この省令は、被験者の人権の保護、安全の保持及び福祉の向上を図り、治験の科学的な質及び成績の信頼性を確保するため、医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律(昭和三十五年法律第百四十五号。以下「法」という。)第十四条第三項及び第十二項(同条第十五項及び法第十九条の二第五項において準用する場合並びに法第十四条の二の二第五項(法第十九条の二第五項において準用する場合を含む。以下同じ。)において読み替えて適用する場合を含む。以下同じ。)並びに法第十四条の四第五項及び第十四条の六第四項(これらの規定を法第十九条の四において準用する場合を含む。以下同じ。)の厚生労働省令で定める基準のうち医薬品の臨床試験の実施に係るもの並びに法第八十条の二第一項、第四項及び第五項に規定する厚生労働省令で定める基準を定めるものとする。
(定義)
第二条 この省令において「製造販売後臨床試験」とは、医薬品の製造販売後の調査及び試験の実施の基準に関する省令(平成十六年厚生労働省令第百七十一号)第二条第一項第三号に規定する製造販売後臨床試験をいう。
2 この省令において「実施医療機関」とは、治験又は製造販売後臨床試験を行う医療機関をいう。
3 この省令において「治験責任医師」とは、実施医療機関において治験に係る業務を統括する医師又は歯科医師をいう。
4 この省令において「製造販売後臨床試験責任医師」とは、実施医療機関において製造販売後臨床試験に係る業務を統括する医師又は歯科医師をいう。
5 この省令において「被験薬」とは、治験の対象とされる薬物又は製造販売後臨床試験の対象とされる医薬品をいう。
6 この省令において「対照薬」とは、治験又は製造販売後臨床試験において被験薬と比較する目的で用いられる薬物をいう。
7 この省令において「治験薬」とは、被験薬及び対照薬(治験に係るものに限る。)をいう。
8 この省令において「製造販売後臨床試験薬」とは、被験薬及び対照薬(製造販売後臨床試験に係るものに限る。)をいう。
9 この省令において「治験使用薬」とは、被験薬(治験に係るものに限る。以下この項において同じ。)並びに被験薬の有効性及び安全性の評価のために使用する薬物をいう。
10 この省令において「治験使用薬等」とは、治験使用薬又は治験使用薬と成分が同一性を有すると認められる薬物をいう。
11 この省令において「製造販売後臨床試験使用薬」とは、被験薬(製造販売後臨床試験に係るものに限る。以下この項において同じ。)並びに被験薬の有効性及び安全性の評価のために使用する薬物をいう。
12 この省令において「製造販売後臨床試験使用薬等」とは、製造販売後臨床試験使用薬又は製造販売後臨床試験使用薬と成分が同一性を有すると認められる薬物をいう。
13 この省令において「被験者」とは、治験薬若しくは製造販売後臨床試験薬を投与される者又は当該者の対照とされる者をいう。
14 この省令において「原資料」とは、被験者に対する治験薬又は製造販売後臨床試験薬の投与及び診療により得られたデータその他の記録をいう。
15 この省令において「治験分担医師」とは、実施医療機関において、治験責任医師の指導の下に治験に係る業務を分担する医師又は歯科医師をいう。
16 この省令において「製造販売後臨床試験分担医師」とは、実施医療機関において、製造販売後臨床試験責任医師の指導の下に製造販売後臨床試験に係る業務を分担する医師又は歯科医師をいう。
17 この省令において「症例報告書」とは、原資料のデータ及びそれに対する治験責任医師若しくは治験分担医師又は製造販売後臨床試験責任医師若しくは製造販売後臨床試験分担医師の評価を被験者ごとに記載した文書をいう。
18 この省令において「治験協力者」とは、実施医療機関において、治験責任医師又は治験分担医師の指導の下にこれらの者の治験に係る業務に協力する薬剤師、看護師その他の医療関係者をいう。
19 この省令において「製造販売後臨床試験協力者」とは、実施医療機関において、製造販売後臨床試験責任医師又は製造販売後臨床試験分担医師の指導の下にこれらの者の製造販売後臨床試験に係る業務に協力する薬剤師、看護師その他の医療関係者をいう。
20 この省令において「治験調整医師」とは、一の治験実施計画書(第二十二項に規定する治験実施計画書をいう。以下この項及び次項において同じ。)に基づき複数の実施医療機関において治験を行う場合に、治験依頼者(第二十二項に規定する治験依頼者をいう。次項において同じ。)又は自ら治験を実施する者により当該実施医療機関における当該治験実施計画書の解釈その他の治験の細目について調整する業務(以下この条において「調整業務」という。)の委嘱を受け、当該調整業務を行う医師又は歯科医師をいう。
21 この省令において「治験調整委員会」とは、一の治験実施計画書に基づき複数の実施医療機関において治験を行う場合に、治験依頼者又は自ら治験を実施する者により調整業務の委嘱を受けて当該調整業務を行う複数の医師又は歯科医師で構成される委員会をいう。
22 この省令において「モニタリング」とは、治験又は製造販売後臨床試験が適正に行われることを確保するため、治験又は製造販売後臨床試験の進捗状況並びに治験又は製造販売後臨床試験がこの省令及び治験の計画書(以下「治験実施計画書」という。)又は製造販売後臨床試験の計画書(以下「製造販売後臨床試験実施計画書」という。)に従って行われているかどうかについて治験の依頼をした者(以下「治験依頼者」という。)若しくは製造販売後臨床試験の依頼をした者(以下「製造販売後臨床試験依頼者」という。)が実施医療機関に対して行う調査又は自ら治験を実施する者が実施医療機関に対して特定の者を指定して行わせる調査をいう。
23 この省令において「監査」とは、治験又は製造販売後臨床試験により収集された資料の信頼性を確保するため、治験又は製造販売後臨床試験がこの省令及び治験実施計画書又は製造販売後臨床試験実施計画書に従って行われたかどうかについて治験依頼者若しくは製造販売後臨床試験依頼者が行う調査、又は自ら治験を実施する者が特定の者を指定して行わせる調査をいう。
24 この省令において「有害事象」とは、治験使用薬又は製造販売後臨床試験使用薬を投与された被験者に生じた全ての疾病又はその徴候をいう。
25 この省令において「代諾者」とは、被験者の親権を行う者、配偶者、後見人その他これらに準じる者をいう。
26 この省令において「自ら治験を実施しようとする者」とは、その所属する実施医療機関等において自ら治験を実施するために法第八十条の二第二項の規定に基づき治験の計画を届け出ようとする者であって、治験責任医師となるべき医師又は歯科医師(一の治験実施計画書に基づき複数の実施医療機関において共同で治験を行う場合にあっては、代表して同項の規定に基づき治験の計画を届け出ようとする治験調整医師となるべき医師又は歯科医師を含む。)をいう。
27 この省令において「自ら治験を実施する者」とは、その所属する実施医療機関等において自ら治験を実施するために法第八十条の二第二項の規定に基づき治験の計画を届け出た治験責任医師(一の治験実施計画書に基づき複数の実施医療機関において共同で治験を行う場合にあっては、代表して同項の規定に基づき治験の計画を届け出た治験調整医師を含む。)をいう。
28 この省令において「治験薬提供者」とは、自ら治験を実施する者に対して治験薬を提供する者をいう。
29 この省令において「拡大治験」とは、人道的見地から実施される治験をいう。
(承認審査資料の基準)
第三条 法第十四条第一項若しくは第十五項(法第十九条の二第五項において準用する場合を含む。)又は第十九条の二第一項の承認を受けようとする者が行う医薬品の臨床試験の実施に係る法第十四条第三項及び第十二項に規定する資料の収集及び作成については、第二章第一節、第三章第一節及び第四章(第二十九条第一項第二号、第三十一条第四項、第三十二条第四項及び第七項、第三十三条第三項並びに第四十八条第三項を除く。)の規定の定めるところによる。
2 自ら治験を実施する者が行う医薬品の臨床試験の実施に係る法第十四条第三項及び第十二項に規定する資料の収集及び作成については、第二章第二節、第三章第二節及び第四章(第二十九条第一項第一号、第三十二条第六項及び第八項並びに第四十八条第二項を除く。)の規定の定めるところによる。
第二章 治験の準備に関する基準
第一節 治験の依頼をしようとする者による治験の準備に関する基準
(業務手順書等)
第四条 治験の依頼をしようとする者は、治験実施計画書の作成、実施医療機関及び治験責任医師の選定、治験使用薬の管理、治験使用薬等の副作用情報等の収集、記録の保存その他の治験の依頼及び管理に係る業務に関する手順書を作成しなければならない。
2 治験の依頼をしようとする者は、医師、歯科医師、薬剤師その他の治験の依頼及び管理に係る業務を行うことにつき必要な専門的知識を有する者を確保しなければならない。
(毒性試験等の実施)
第五条 治験の依頼をしようとする者は、被験薬の品質、毒性及び薬理作用に関する試験その他治験の依頼をするために必要な試験を終了していなければならない。
(実施医療機関等の選定)
第六条 治験の依頼をしようとする者は、第三十五条各号に掲げる要件を満たしている実施医療機関及び第四十二条各号に掲げる要件を満たしている治験責任医師を選定しなければならない。
(治験実施計画書)
第七条 治験の依頼をしようとする者は、次に掲げる事項を記載した治験実施計画書を作成しなければならない。
一 治験の依頼をしようとする者の氏名(法人にあっては、その名称。以下この号及び次号、第十三条第一項第二号及び第三号、第十五条の四第一項第二号及び第六号並びに第十六条第一項第二号において同じ。)及び住所(法人にあっては、その主たる事務所の所在地。以下この号及び次号、第十三条第一項第二号及び第三号、第十五条、第十五条の四第一項第二号及び第六号、第十六条第一項第二号並びに第二十六条第二項において同じ。)(当該者が本邦内に住所を有しない場合にあっては、その氏名及び住所地の国名並びに第十五条に規定する治験国内管理人の氏名及び住所。第十三条第一項第二号において同じ。)
二 治験に係る業務の全部又は一部を委託する場合にあっては、当該業務を受託した者(以下この章において「受託者」という。)の氏名、住所及び当該委託に係る業務の範囲
三 実施医療機関の名称及び所在地
四 治験責任医師となるべき者の氏名
五 治験の目的
六 治験使用薬の概要
七 治験の方法
八 被験者の選定に関する事項
九 原資料の閲覧に関する事項
十 記録(データを含む。)の保存に関する事項
十一 治験調整医師に委嘱した場合にあっては、その氏名
十二 治験調整委員会に委嘱した場合にあっては、これを構成する医師又は歯科医師の氏名
十三 第十九条に規定する効果安全性評価委員会を設置したときは、その旨
2 治験の依頼をしようとする者は、当該治験が被験者に対して治験薬の効果を有しないこと及び第五十条第一項の同意を得ることが困難な者を対象にすることが予測される場合には、その旨及び次に掲げる事項を治験実施計画書に記載しなければならない。
一 当該治験が第五十条第一項の同意を得ることが困難と予測される者を対象にしなければならないことの説明
二 当該治験において、予測される被験者への不利益が必要な最小限度のものであることの説明
3 治験の依頼をしようとする者は、当該治験が第五十条第一項及び第二項の同意を得ることが困難と予測される者を対象にしている場合には、その旨及び次に掲げる事項を治験実施計画書に記載しなければならない。
一 当該被験薬が、生命が危険な状態にある傷病者に対して、その生命の危険を回避するため緊急に使用される医薬品として、製造販売の承認を申請することを予定しているものであることの説明
二 現在における治療方法では被験者となるべき者に対して十分な効果が期待できないことの説明
三 被験薬の使用により被験者となるべき者の生命の危険が回避できる可能性が十分にあることの説明
四 第十九条に規定する効果安全性評価委員会が設置されている旨
4 第一項の規定により治験実施計画書を作成するときは、当該治験実施計画書の内容及びこれに従って治験を行うことについて、治験責任医師となるべき者の同意を得なければならない。
5 治験の依頼をしようとする者は、治験使用薬の品質、有効性及び安全性に関する事項その他の治験を適正に行うために重要な情報を知ったときは、必要に応じ、当該治験実施計画書を改訂しなければならない。この場合においては、前項の規定を準用する。
(治験薬概要書)
第八条 治験の依頼をしようとする者は、第五条の試験により得られた資料並びに被験薬の品質、有効性及び安全性に関する情報に基づいて、次に掲げる事項を記載した治験薬概要書を作成しなければならない。
一 被験薬の化学名又は識別記号
二 品質、毒性、薬理作用その他の被験薬に関する事項
三 臨床試験が実施されている場合にあっては、その試験成績に関する事項
2 治験の依頼をしようとする者は、被験薬の品質、有効性及び安全性に関する事項その他の治験を適正に行うために重要な情報を知ったときは、必要に応じ、前項の治験薬概要書を改訂しなければならない。
(説明文書の作成の依頼)
第九条 治験の依頼をしようとする者は、治験責任医師となるべき者に対して、第五十条第一項の規定により説明を行うために用いられる文書(以下「説明文書」という。)の作成を依頼しなければならない。
(実施医療機関の長への文書の事前提出)
第十条 治験の依頼をしようとする者は、あらかじめ、次に掲げる文書を実施医療機関の長に提出しなければならない。
一 治験実施計画書(第七条第五項の規定により改訂されたものを含む。)
二 治験薬概要書(第八条第二項の規定により改訂されたものを含む。)及び治験使用薬(被験薬を除く。)に係る科学的知見を記載した文書
三 症例報告書の見本
四 説明文書
五 治験責任医師及び治験分担医師(以下「治験責任医師等」という。)となるべき者の氏名を記載した文書
六 治験の費用の負担について説明した文書
七 被験者の健康被害の補償について説明した文書
2 治験の依頼をしようとする者は、前項の規定による文書の提出に代えて、第四項で定めるところにより、当該実施医療機関の長の承諾を得て、前項各号に掲げる文書に記載すべき事項を電子情報処理組織を使用する方法その他の情報通信の技術を利用する方法であって次に掲げるもの(以下「電磁的方法」という。)により提出することができる。この場合において、当該治験の依頼をしようとする者は、当該文書を提出したものとみなす。
一 治験の依頼をしようとする者の使用に係る電子計算機と、実施医療機関の長の使用に係る電子計算機とを電気通信回線で接続した電子情報処理組織を使用する方法のうちイ又はロに掲げるもの
イ 治験の依頼をしようとする者の使用に係る電子計算機と実施医療機関の長の使用に係る電子計算機とを接続する電気通信回線を通じて送信し、受信者の使用に係る電子計算機に備えられたファイルに記録する方法
ロ 治験の依頼をしようとする者の使用に係る電子計算機に備えられたファイルに記録された前項各号に掲げる事項を電気通信回線を通じて実施医療機関の長の閲覧に供し、当該実施医療機関の長の使用に係る電子計算機に備えられたファイルに同項各号に掲げる事項を記録する方法(電磁的方法による文書の提出を受ける旨の承諾又は受けない旨の申出をする場合にあっては、治験の依頼をしようとする者の使用に係る電子計算機に備えられたファイルにその旨を記録する方法)
二 磁気ディスク、シー・ディー・ロムその他これらに準ずる方法により一定の事項を確実に記録しておくことができる物をもって調製するファイルに前項各号に掲げる事項を記録したものを交付する方法
3 前項各号に掲げる方法は、実施医療機関の長がファイルへの記録を出力することにより書面を作成することができるものでなければならない。
4 治験の依頼をしようとする者は、第二項の規定により第一項各号に掲げる文書を提出しようとするときは、あらかじめ、当該実施医療機関の長に対し、その用いる次に掲げる電磁的方法の種類及び内容を示し、書面又は電磁的方法による承諾を得なければならない。
一 第二項各号に規定する方法のうち治験の依頼をしようとする者が使用するもの
二 ファイルへの記録の方式
5 前項の承諾を得た治験の依頼をしようとする者は、当該実施医療機関の長から書面又は電磁的方法により電磁的方法による通知を受けない旨の申出があったときは、当該実施医療機関の長に対し、第一項各号に掲げる文書の提出を電磁的方法によってしてはならない。ただし、当該実施医療機関の長が再び前項の承諾をした場合は、この限りでない。
(治験薬の事前交付の禁止)
第十一条 治験の依頼をしようとする者は、治験の契約が締結される前に、実施医療機関に対して治験薬を交付してはならない。
(業務の委託)
第十二条 治験の依頼をしようとする者は、治験の依頼及び管理に係る業務の全部又は一部を委託する場合には、次に掲げる事項を記載した文書により当該委託を受けた者(以下この節において「受託者」という。)との契約を締結しなければならない。
一 当該委託に係る業務の範囲
二 当該委託に係る業務の手順に関する事項
三 前号の手順に基づき当該委託に係る業務が適正かつ円滑に行われているかどうかを治験の依頼をしようとする者が確認することができる旨
四 受託者に対する指示に関する事項
五 前号の指示を行った場合において当該措置が講じられたかどうかを治験の依頼をしようとする者が確認することができる旨
六 受託者が治験の依頼をしようとする者に対して行う報告に関する事項
七 当該委託する業務に係る第十四条の措置に関する事項
八 その他当該委託に係る業務について必要な事項
2 治験の依頼をしようとする者は、前項の規定による文書による契約の締結に代えて、第四項で定めるところにより、前項の受託者の承諾を得て、前項各号に掲げる事項を内容とする契約を電子情報処理組織を使用する方法その他の情報通信の技術を利用する方法であって次に掲げるもの(以下この条において「電磁的方法」という。)により締結することができる。この場合において、当該治験の依頼をしようとする者は、当該文書による契約を締結したものとみなす。
一 治験の依頼をしようとする者の使用に係る電子計算機と、受託者の使用に係る電子計算機とを電気通信回線で接続した電子情報処理組織を使用する方法のうちイ又はロに掲げるもの
イ 治験の依頼をしようとする者の使用に係る電子計算機と受託者の使用に係る電子計算機とを接続する電気通信回線を通じて送信し、それぞれの使用に係る電子計算機に備えられたファイルに記録する方法
ロ 治験の依頼をしようとする者の使用に係る電子計算機に備えられたファイルに記録された前項各号に掲げる事項を電気通信回線を通じて受託者の閲覧に供し、当該受託者の使用に係る電子計算機に備えられたファイルに同項各号に掲げる事項を記録する方法(電磁的方法による契約の締結を行う旨の承諾又は行わない旨の申出をする場合にあっては、治験の依頼をしようとする者の使用に係る電子計算機に備えられたファイルにその旨を記録する方法)
二 磁気ディスク、シー・ディー・ロムその他これらに準ずる方法により一定の事項を確実に記録しておくことができる物をもって調製するファイルに前項各号に掲げる事項を記録したものを交付する方法
3 前項各号に掲げる方法は、次に掲げる技術的基準に適合するものでなければならない。
一 治験の依頼をしようとする者及び受託者がファイルへの記録を出力することにより書面を作成することができるものであること。
二 ファイルに記録された文書に記載すべき事項について、改変が行われていないかどうかを確認することができる措置を講じていること。
4 治験の依頼をしようとする者は、第二項の規定により第一項各号に掲げる事項を内容とする契約を締結しようとするときは、あらかじめ、当該受託者に対し、その用いる次に掲げる電磁的方法の種類及び内容を示し、書面又は電磁的方法により承諾を得なければならない。
一 第二項各号に掲げる方法のうち治験の依頼をしようとする者が使用するもの
二 ファイルへの記録の方式
5 前項の規定による承諾を得た治験の依頼をしようとする者は、受託者から書面又は電磁的方法により電磁的方法による契約を締結しない旨の申出があったときは、受託者に対し、第一項各号に掲げる事項を内容とする契約の締結を電磁的方法によってしてはならない。ただし、受託者が再び前項の規定による承諾をした場合は、この限りでない。
(治験の契約)
第十三条 治験の依頼をしようとする者及び実施医療機関(前条の規定により業務の全部又は一部を委託する場合にあっては、治験の依頼をしようとする者、受託者及び実施医療機関)は、次に掲げる事項について記載した文書により治験の契約を締結しなければならない。
一 契約を締結した年月日
二 治験の依頼をしようとする者の氏名及び住所
三 前条の規定により業務の全部又は一部を委託する場合にあっては、受託者の氏名、住所及び当該委託した業務の範囲
四 実施医療機関の名称及び所在地
五 契約担当者の氏名及び職名
六 治験責任医師の氏名
七 治験の期間
八 治験使用薬の管理に関する事項
九 記録(データを含む。)の保存に関する事項
十 この省令の規定により治験依頼者及び実施医療機関に従事する者が行う通知に関する事項
十一 被験者の秘密の保全に関する事項
十二 治験の費用に関する事項
十三 実施医療機関が治験実施計画書を遵守して治験を行う旨
十四 実施医療機関が治験依頼者の求めに応じて第四十一条第二項各号に掲げる記録(文書を含む。)を閲覧に供する旨
十五 実施医療機関がこの省令、治験実施計画書又は当該契約に違反することにより適正な治験に支障を及ぼしたと認める場合(第四十六条に規定する場合を除く。)には、治験依頼者が治験の契約を解除できる旨
十六 被験者の健康被害の補償に関する事項
十七 その他治験が適正かつ円滑に行われることを確保するために必要な事項
2 前項の文書による契約については、前条第二項から第五項までの規定を準用する。この場合において、同条第二項中「前項の受託者」とあるのは「実施医療機関(この条の規定により業務の全部又は一部を委託する場合にあっては、実施医療機関及び受託者)(以下「実施医療機関等」という。)」と、同項第一号並びに同条第三項第一号、第四項及び第五項中「受託者」とあるのは「実施医療機関等」と読み替えるものとする。
(被験者に対する補償措置)
第十四条 治験の依頼をしようとする者は、あらかじめ、治験に係る被験者に生じた健康被害(受託者の業務により生じたものを含む。)の補償のために、保険契約の締結その他の必要な措置を講じておかなければならない。
(治験国内管理人)
第十五条 本邦内に住所を有しない治験の依頼をしようとする者は、治験使用薬による保健衛生上の危害の発生又は拡大の防止に必要な措置を採らせるため、治験の依頼をしようとする者に代わって治験の依頼を行うことができる者を、本邦内に住所を有する者(外国法人で本邦内に事務所を有するものの当該事務所の代表者を含む。)のうちから選任し、この者(以下「治験国内管理人」という。)に治験の依頼に係る手続を行わせなければならない。
第二節 自ら治験を実施しようとする者による治験の準備に関する基準
(業務手順書等)
第十五条の二 自ら治験を実施しようとする者は、治験実施計画書の作成、治験使用薬の管理、治験使用薬等の副作用情報等の収集、記録の保存その他の治験の実施の準備及び管理に係る業務に関する手順書を作成しなければならない。
2 自ら治験を実施しようとする者は、医師、歯科医師、薬剤師その他の治験の実施の準備及び管理に係る業務を行うことにつき必要な専門的知識を有する者を確保しなければならない。
(毒性試験等の実施)
第十五条の三 自ら治験を実施しようとする者は、被験薬の品質、毒性及び薬理作用に関する試験その他治験を実施するために必要な試験を終了していなければならない。
(治験実施計画書)
第十五条の四 自ら治験を実施しようとする者は、次に掲げる事項を記載した治験実施計画書を作成しなければならない。
一 自ら治験を実施しようとする者の氏名及び住所
二 治験の実施の準備及び管理に係る業務の全部又は一部を委託する場合にあっては、当該受託者の氏名、住所及び当該委託に係る業務の範囲
三 実施医療機関の名称及び所在地
四 治験の目的
五 治験使用薬の概要
六 治験薬提供者の氏名及び住所
七 治験の方法
八 被験者の選定に関する事項
九 原資料の閲覧に関する事項
十 記録(データを含む。)の保存に関する事項
十一 治験調整医師に委嘱した場合にあっては、その氏名
十二 治験調整委員会に委嘱した場合にあっては、これを構成する医師又は歯科医師の氏名
十三 第二十六条の五に規定する効果安全性評価委員会を設置したときは、その旨
2 自ら治験を実施しようとする者は、当該治験が被験者に対して治験薬の効果を有しないこと及び第五十条第一項の同意を得ることが困難な者を対象にすることが予測される場合には、その旨及び次に掲げる事項を治験実施計画書に記載しなければならない。
一 当該治験が第五十条第一項の同意を得ることが困難と予測される者を対象にしなければならないことの説明
二 当該治験において、予測される被験者への不利益が必要な最小限度のものであることの説明
3 自ら治験を実施しようとする者は、当該治験が第五十条第一項及び第二項の同意を得ることが困難と予測される者を対象にしている場合には、その旨及び次に掲げる事項を治験実施計画書に記載しなければならない。
一 当該被験薬が、生命が危険な状態にある傷病者に対して、その生命の危険を回避するため緊急に使用される医薬品として、製造販売の承認を申請することを予定しているものであることの説明
二 現在における治療方法では被験者となるべき者に対して十分な効果が期待できないことの説明
三 被験薬の使用により被験者となるべき者の生命の危険が回避できる可能性が十分にあることの説明
四 第二十六条の五に規定する効果安全性評価委員会が設置されている旨
4 自ら治験を実施しようとする者は、治験使用薬の品質、有効性及び安全性に関する事項その他の治験を適正に行うために重要な情報を知ったときは、必要に応じ、治験実施計画書を改訂しなければならない。
(治験薬概要書)
第十五条の五 自ら治験を実施しようとする者は、第十五条の三の試験により得られた資料並びに被験薬の品質、有効性及び安全性に関する情報に基づいて、次に掲げる事項を記載した治験薬概要書を作成しなければならない。
一 被験薬の化学名又は識別記号
二 品質、毒性、薬理作用その他の被験薬に関する事項
三 臨床試験が実施されている場合にあっては、その試験成績に関する事項
2 自ら治験を実施しようとする者は、被験薬の品質、有効性及び安全性に関する事項その他の治験を適正に行うために重要な情報を知ったときは、必要に応じ、前項の治験薬概要書を改訂しなければならない。
(説明文書の作成)
第十五条の六 自ら治験を実施しようとする者(治験責任医師となるべき医師又は歯科医師に限る。次条及び第二十六条の四において同じ。)は、説明文書を作成しなければならない。
(実施医療機関の長への文書の事前提出等)
第十五条の七 自ら治験を実施しようとする者は、あらかじめ、次に掲げる文書を実施医療機関の長に提出し、治験の実施の承認を得なければならない。
一 治験実施計画書(第十五条の四第四項の規定により改訂されたものを含む。)
二 治験薬概要書(第十五条の五第二項の規定により改訂されたものを含む。)及び治験使用薬(被験薬を除く。)に係る科学的知見を記載した文書
三 症例報告書の見本
四 説明文書
五 モニタリングに関する手順書
六 監査に関する計画書及び業務に関する手順書
七 治験分担医師となるべき者の氏名を記載した文書
八 治験使用薬の管理に関する事項を記載した文書
九 この省令の規定により自ら治験を実施する者及び実施医療機関に従事する者が行う通知に関する事項を記載した文書
十 治験の費用に関する事項を記載した文書
十一 被験者の健康被害の補償に関する事項を記載した文書
十二 実施医療機関が自ら治験を実施する者の求めに応じて第四十一条第二項各号に掲げる記録(文書を含む。)を閲覧に供する旨を記載した文書
十三 実施医療機関がこの省令又は治験実施計画書に違反することにより適正な治験に支障を及ぼしたと認める場合(第四十六条に規定する場合を除く。)には、自ら治験を実施する者は治験を中止することができる旨を記載した文書
十四 その他治験が適正かつ円滑に行われることを確保するために必要な事項を記載した文書
(業務の委託)
第十五条の八 自ら治験を実施しようとする者又は実施医療機関は、治験の実施の準備及び管理に係る業務の全部又は一部を委託する場合には、次に掲げる事項を記載した文書により当該委託を受けた者(以下この節において「受託者」という。)との契約を締結しなければならない。
一 当該委託に係る業務の範囲
二 当該委託に係る業務の手順に関する事項
三 前号の手順に基づき当該委託に係る業務が適正かつ円滑に行われているかどうかを自ら治験を実施しようとする者又は実施医療機関が確認することができる旨
四 受託者に対する指示に関する事項
五 前号の指示を行った場合において当該措置が講じられたかどうかを自ら治験を実施しようとする者又は実施医療機関が確認することができる旨
六 受託者が自ら治験を実施しようとする者又は実施医療機関に対して行う報告に関する事項
七 当該委託する業務に係る次条に規定する措置に関する事項
八 その他当該委託に係る業務について必要な事項
2 前項の規定による文書の契約の締結については、第十二条第二項から第五項までの規定を準用する。この場合において、これらの規定中「治験の依頼をしようとする者」とあるのは「自ら治験を実施しようとする者又は実施医療機関」と読み替えるものとする。
(被験者に対する補償措置)
第十五条の九 自ら治験を実施しようとする者は、あらかじめ、治験に係る被験者に生じた健康被害(受託者の業務により生じたものを含む。)の補償のために、保険契約の締結その他の必要な措置を講じておかなければならない。
第三章 治験の管理に関する基準
第一節 治験依頼者による治験の管理に関する基準
(治験薬又は治験使用薬の管理)
第十六条 治験依頼者は、治験薬の容器又は被包に次に掲げる事項(拡大治験を実施する場合にあっては、第一号及び第二号に掲げる事項に限る。)を邦文で記載しなければならない。
一 治験用である旨
二 治験依頼者の氏名及び住所(当該者が本邦内に住所を有しない場合にあっては、その氏名及び住所地の国名並びに治験国内管理人の氏名及び住所)
三 化学名又は識別記号
四 製造番号又は製造記号
五 貯蔵方法、有効期間等を定める必要があるものについては、その内容
2 治験依頼者は、治験薬に添付する文書、その治験薬又はその容器若しくは被包(内袋を含む。)には、次に掲げる事項を記載してはならない。ただし、被験者、治験責任医師等若しくは治験協力者が被験薬及び対照薬の識別をできない状態にしていない治験薬を用いる治験又は拡大治験を実施する場合にあっては、この限りでない。
一 予定される販売名
二 予定される効能又は効果
三 予定される用法又は用量
3 治験依頼者は、被験者、治験責任医師等及び治験協力者が被験薬及び対照薬の識別をできない状態で実施医療機関に交付した治験薬について、緊急時に、治験責任医師等が被験薬及び対照薬の識別を直ちにできるよう必要な措置を講じておかなければならない。
4 治験依頼者は、輸送及び保存中の汚染や劣化を防止するため治験薬を包装して実施医療機関に交付しなければならない。
5 治験依頼者は、治験薬又は治験使用薬に関する次に掲げる記録を作成しなければならない。
一 治験薬の製造年月日、製造方法、製造数量等の製造に関する記録及び治験薬の安定性等の品質に関する試験の記録
二 実施医療機関ごとの治験使用薬の交付又は回収の数量及び年月日の記録
三 治験使用薬の処分の記録
6 治験依頼者は、治験の契約の締結後遅滞なく、実施医療機関における治験使用薬の管理に関する手順書を作成し、これを実施医療機関に交付しなければならない。
7 治験依頼者は、必要に応じ、治験薬の溶解方法その他の取扱方法を説明した文書を作成し、これを治験責任医師等、治験協力者及び第三十九条に規定する治験薬管理者に交付しなければならない。
8 第六項の規定による手順書の交付については、第十条第二項から第五項までの規定を準用する。この場合において、これらの規定中「治験の依頼をしようとする者」とあるのは、「治験依頼者」と読み替えるものとする。
9 第七項の文書の交付については、第十条第二項から第五項までの規定を準用する。この場合において、これらの規定中「治験の依頼をしようとする者」とあるのは「治験依頼者」と、「実施医療機関の長」とあるのは「治験責任医師等、治験協力者及び第三十九条に規定する治験薬管理者」と読み替えるものとする。
(治験薬の交付)
第十七条 治験依頼者は、治験薬の品質の確保のために必要な構造設備を備え、かつ、適切な製造管理及び品質管理の方法が採られている製造所において製造された治験薬を、治験依頼者の責任のもと実施医療機関に交付しなければならない。ただし、拡大治験を実施する場合にあっては、実施医療機関が在庫として保管する医薬品の中から、治験薬として使用する医薬品を当該実施医療機関に選定させること又は治験依頼者自ら選定することができる。
2 治験依頼者は、前項ただし書の場合には、適切な製造管理及び品質管理の方法が採られている場所において、治験薬の容器又は被包に前条第一項第一号及び第二号に掲げる事項を邦文で記載しなければならない。
3 第三十九条に規定する治験薬管理者は、第一項ただし書の場合には、当該治験薬とそれ以外の医薬品とを区別して適切に管理しなければならない。
(委嘱の文書の作成)
第十八条 治験依頼者は、第二条第二十項に規定する調整業務を治験調整医師又は治験調整委員会に委嘱する場合には、その業務の範囲、手順その他必要な事項を記載した文書を作成しなければならない。
(効果安全性評価委員会の設置)
第十九条 治験依頼者は、治験の継続の適否又は治験実施計画書の変更について審議させるために効果安全性評価委員会を設置することができる。
2 治験依頼者は、前項の効果安全性評価委員会の審議に関する手順書を作成し、これに従って審議を行わせなければならない。
3 治験依頼者は、前項の審議を行ったときは、その審議の記録を作成し、これを保存しなければならない。
(副作用情報等)
第二十条 治験依頼者は、治験使用薬の品質、有効性及び安全性に関する事項その他の治験を適正に行うために必要な情報を収集し、及び検討するとともに、実施医療機関の長に対し、これを提供しなければならない。
2 治験依頼者は、治験使用薬について法第八十条の二第六項に規定する事項を知ったときは、その発現症例一覧等を当該被験薬ごとに、当該被験薬について初めて治験の計画を届け出た日等から起算して一年ごとに、その期間の満了後三月以内に治験責任医師及び実施医療機関の長に通知しなければならない。
3 治験依頼者は、前項に規定する事項のうち当該被験薬の治験薬概要書又は治験使用薬(被験薬を除く。)に係る科学的知見から予測できないものを知ったときは、直ちにその旨を治験責任医師及び実施医療機関の長に通知しなければならない。
4 治験依頼者は、治験使用薬の品質、有効性及び安全性に関する事項その他の治験を適正に行うために重要な情報を知ったときは、必要に応じ、治験実施計画書及び治験薬概要書を改訂しなければならない。この場合において、治験実施計画書の改訂について治験責任医師の同意を得なければならない。
(モニタリングの実施)
第二十一条 治験依頼者は、モニタリングに関する手順書を作成し、当該手順書に従ってモニタリングを実施しなければならない。
2 前項の規定によりモニタリングを実施する場合には、実施医療機関において実地に行わなければならない。ただし、他の方法により十分にモニタリングを実施することができる場合には、この限りではない。
(モニターの責務)
第二十二条 モニタリングに従事する者(以下「モニター」という。)は、モニタリングの結果、実施医療機関における治験がこの省令又は治験実施計画書に従って行われていないことを確認した場合には、その旨を直ちに当該実施医療機関の治験責任医師に告げなければならない。
2 モニターは、モニタリングの実施の際、実施医療機関において実地に行い、又はこれと連絡を取ったときは、その都度次に掲げる事項を記載したモニタリング報告書を治験依頼者に提出しなければならない。
一 モニタリングを行った日付
二 モニタリングの対象となった実施医療機関
三 モニターの氏名
四 モニタリングの際に説明等を聴取した治験責任医師等の氏名
五 モニタリングの結果の概要
六 前項の規定により治験責任医師に告げた事項
七 前号の事項について講じられるべき措置及び当該措置に関するモニターの所見
(監査)
第二十三条 治験依頼者は、監査に関する計画書及び業務に関する手順書を作成し、当該計画書及び手順書に従って監査を実施しなければならない。
2 監査に従事する者(以下「監査担当者」という。)は、監査に係る医薬品の開発に係る部門及びモニタリングを担当する部門に属してはならない。
3 監査担当者は、監査を実施した場合には、監査で確認した事項を記録した監査報告書及び監査が実施されたことを証明する監査証明書を作成し、これを治験依頼者に提出しなければならない。
(治験の中止等)
第二十四条 治験依頼者は、実施医療機関がこの省令、治験実施計画書又は治験の契約に違反することにより適正な治験に支障を及ぼしたと認める場合(第四十六条に規定する場合を除く。)には、当該実施医療機関との治験の契約を解除し、当該実施医療機関における治験を中止しなければならない。
2 治験依頼者は、治験を中断し、又は中止する場合には、速やかにその旨及びその理由を実施医療機関の長に文書により通知しなければならない。
3 治験依頼者は、当該治験により収集された臨床試験の試験成績に関する資料を法第十四条第三項及び医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律施行規則(昭和三十六年厚生省令第一号)第四十五条の四第一項に規定する申請書に添付しないことを決定した場合には、その旨及びその理由を実施医療機関の長に文書により通知しなければならない。
4 第二項及び前項の規定による文書による通知については、第十条第二項から第五項までの規定を準用する。この場合において、これらの規定中「治験の依頼をしようとする者」とあるのは、「治験依頼者」と読み替えるものとする。
(総括報告書)
第二十五条 治験依頼者は、治験を終了し、又は中止したときは、総括報告書(治験の結果等を取りまとめた文書をいう。以下同じ。)を作成しなければならない。
(記録の保存等)
第二十六条 治験依頼者は、次に掲げる治験に関する記録(文書及びデータを含む。)を被験薬に係る医薬品についての製造販売の承認(法第十四条の二の二第一項の規定により条件及び期限を付したものを除く。第二十六条の十二、第三十四条及び第四十一条第二項において同じ。)を受ける日(第二十四条第三項の規定により通知したときは、通知した日後三年を経過した日)又は治験の中止若しくは終了の後三年を経過した日のうちいずれか遅い日までの期間適切に保存しなければならない。
一 治験実施計画書、契約書、総括報告書その他この省令の規定により治験依頼者が作成した文書又はその写し
二 症例報告書、第三十二条第六項の規定により通知された文書その他この省令の規定により実施医療機関の長又は治験責任医師等から入手した記録
三 モニタリング、監査その他の治験の依頼及び管理に係る業務の記録(前二号及び第五号に掲げるものを除く。)
四 治験を行うことにより得られたデータ
五 第十六条第五項の記録
2 本邦内に住所を有しない治験依頼者は、治験国内管理人に第十六条第五項の記録を前項に定める期間保存させなければならない。
第二節 自ら治験を実施する者による治験の管理に関する基準
(治験薬又は治験使用薬の管理)
第二十六条の二 自ら治験を実施する者は、治験薬の容器又は被包に次に掲げる事項(拡大治験を実施する場合にあっては、第一号及び第二号に掲げる事項に限る。)を邦文で記載しなければならない。
一 治験用である旨
二 自ら治験を実施する者の氏名及び住所
三 化学名又は識別記号
四 製造番号又は製造記号
五 貯蔵方法、有効期間等を定める必要があるものについては、その内容
2 自ら治験を実施する者は、治験薬に添付する文書、その治験薬又はその容器若しくは被包(内袋を含む。)には、次に掲げる事項を記載してはならない。ただし、被験者、治験責任医師等若しくは治験協力者が被験薬及び対照薬の識別をできない状態にしていない治験薬を用いる治験又は拡大治験を実施する場合にあっては、この限りでない。
一 予定される販売名
二 予定される効能又は効果
三 予定される用法又は用量
3 自ら治験を実施する者は、被験者、治験分担医師及び治験協力者が被験薬及び対照薬の識別をできない状態で入手した治験薬について、緊急時に、治験分担医師が被験薬及び対照薬の識別を直ちにできるよう必要な措置を講じておかなければならない。
4 自ら治験を実施する者は、輸送及び保存中の汚染や劣化を防止するため必要な措置を講じておかなければならない。
5 自ら治験を実施する者は、治験薬又は治験使用薬に関する次に掲げる記録を作成し、又は入手しなければならない。
一 治験薬の製造年月日、製造方法、製造数量等の製造に関する記録及び治験薬の安定性等の品質に関する試験の記録
二 治験使用薬を入手し、又は治験薬提供者から提供を受けた場合にはその数量及び年月日の記録
三 治験使用薬の処分の記録
6 自ら治験を実施する者は、治験の実施の承認後遅滞なく、実施医療機関における治験使用薬の管理に関する手順書を作成し、これを実施医療機関に交付しなければならない。
7 自ら治験を実施する者は、必要に応じ、治験薬の溶解方法その他の取扱方法を説明した文書を作成し、これを治験分担医師、治験協力者及び第三十九条に規定する治験薬管理者に交付しなければならない。
(治験薬の品質の確保)
第二十六条の三 自ら治験を実施する者は、治験薬の品質の確保のために必要な構造設備を備え、かつ、適切な製造管理及び品質管理の方法が採られている製造所において製造された治験薬を用いて治験を実施しなければならない。ただし、拡大治験を実施する場合にあっては、実施医療機関が在庫として保管する医薬品の中から、治験薬として使用する医薬品を当該実施医療機関に選定させること又は自ら治験を実施する者自ら選定することができる。
2 自ら治験を実施する者は、前項ただし書の場合には、適切な製造管理及び品質管理の方法が採られている場所において、治験薬の容器又は被包に前条第一項第一号及び第二号に掲げる事項を邦文で記載しなければならない。
3 第三十九条に規定する治験薬管理者は、第一項ただし書の場合には、当該治験薬とそれ以外の医薬品とを区別して適切に管理しなければならない。
(委嘱の文書の作成)
第二十六条の四 自ら治験を実施する者は、第二条第二十項に規定する調整業務を治験調整医師又は治験調整委員会に委嘱する場合には、その業務の範囲、手順その他必要な事項を記載した文書を作成しなければならない。
(効果安全性評価委員会の設置)
第二十六条の五 自ら治験を実施する者は、治験の継続の適否又は治験実施計画書の変更について審議させるために効果安全性評価委員会を設置することができる。
2 自ら治験を実施する者は、前項の効果安全性評価委員会の審議に関する手順書を作成し、これに従って審議を行わせなければならない。
3 自ら治験を実施する者は、前項の審議を行ったときは、その審議の記録を作成し、これを保存しなければならない。
(副作用情報等)
第二十六条の六 自ら治験を実施する者は、治験使用薬の品質、有効性及び安全性に関する事項その他の治験を適正に行うために必要な情報を収集し、及び検討するとともに、実施医療機関の長に対し、これを提供しなければならない。
2 自ら治験を実施する者は、治験使用薬について法第八十条の二第六項に規定する事項を知ったときは、直ちにその旨を実施医療機関の長(一の実施計画書に基づき共同で複数の実施医療機関において治験を実施する場合には他の実施医療機関の治験責任医師を含む。)に通知しなければならない。
3 自ら治験を実施する者は、治験使用薬の品質、有効性及び安全性に関する事項その他の治験を適正に行うために重要な情報を知ったときは、必要に応じ、治験実施計画書及び治験薬概要書を改訂しなければならない。
(モニタリングの実施)
第二十六条の七 自ら治験を実施する者は、モニタリングに関する手順書を作成し、第二十七条第一項の治験審査委員会の意見を踏まえて、当該手順書に従って、モニタリングを実施させなければならない。
2 モニターは、モニタリングの対象となる実施医療機関においてその対象となる治験に従事してはならない。
3 第一項の規定によりモニタリングを実施する場合には、実施医療機関において実地に行わなければならない。ただし、他の方法により十分にモニタリングを実施することができる場合には、この限りではない。
(モニターの責務)
第二十六条の八 モニターは、モニタリングの結果、実施医療機関における治験がこの省令又は治験実施計画書に従って行われていないことを確認した場合には、その旨を直ちに当該実施医療機関の治験責任医師に告げなければならない。
2 モニターは、モニタリングを実地に実施したときは、その都度次に掲げる事項を記載したモニタリング報告書を自ら治験を実施する者及び当該モニタリングに係る実施医療機関の長に提出しなければならない。
一 モニタリングを行った日付
二 モニターの氏名
三 モニタリングの際に説明等を聴取した治験責任医師等の氏名
四 モニタリングの結果の概要
五 前項の規定により治験責任医師に告げた事項
六 前号の事項について講じられるべき措置及び当該措置に関するモニターの所見
(監査)
第二十六条の九 自ら治験を実施する者は、監査に関する計画書及び業務に関する手順書を作成し、第二十七条第一項の治験審査委員会の意見を踏まえて、当該計画書及び手順書に従って監査を実施させなければならない。
2 監査担当者は、当該監査に係る治験を実施する医療機関において当該治験の実施(その準備及び管理を含む。)及びモニタリングに従事してはならない。
3 監査担当者は、監査を実施した場合には、監査で確認した事項を記録した監査報告書及び監査が実施されたことを証明する監査証明書を作成し、これを自ら治験を実施する者及び実施医療機関の長に提出しなければならない。
(治験の中止等)
第二十六条の十 自ら治験を実施する者は、実施医療機関がこの省令又は治験実施計画書に違反することにより適正な治験に支障を及ぼしたと認める場合(第四十六条に規定する場合を除く。)には、当該実施医療機関における治験を中止しなければならない。
2 自ら治験を実施する者は、治験を中断し、又は中止する場合には、速やかにその旨及びその理由を実施医療機関の長に文書により通知しなければならない。
3 自ら治験を実施する者は、当該治験により収集された臨床試験の試験成績に関する資料が法第十四条第三項及び医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律施行規則第四十五条の四第一項の申請書に添付されないことを知り得た場合には、その旨及びその理由を実施医療機関の長に文書により通知しなければならない。
(総括報告書)
第二十六条の十一 自ら治験を実施する者は、治験を終了し、又は中止したときは、総括報告書を作成しなければならない。
(記録の保存等)
第二十六条の十二 自ら治験を実施する者は、次に掲げる治験に関する記録(文書及びデータを含む。)を、治験薬提供者が被験薬に係る医薬品についての製造販売の承認を受ける日(第二十六条の十第三項の規定により通知したときは、通知した日後三年を経過した日)又は治験の中止若しくは終了の後三年を経過した日のうちいずれか遅い日までの期間適切に保存しなければならない。
一 治験実施計画書、承認書、総括報告書その他この省令の規定により自ら治験を実施する者が作成した文書又はその写し
二 症例報告書、第三十二条第七項の規定により通知された文書その他この省令の規定により実施医療機関の長又は治験分担医師から入手した記録
三 モニタリング、監査その他の治験の実施の基準及び管理に係る業務の記録(前二号及び第五号に掲げるものを除く。)
四 治験を行うことにより得られたデータ
五 第二十六条の二第五項に規定する記録
第四章 治験を行う基準
第一節 治験審査委員会
(治験審査委員会の設置)
第二十七条 実施医療機関の長は、治験を行うことの適否その他の治験に関する調査審議を次に掲げるいずれかの治験審査委員会に行わせなければならない。
一 実施医療機関の長が設置した治験審査委員会
二 一般社団法人又は一般財団法人が設置した治験審査委員会
三 特定非営利活動促進法(平成十年法律第七号)第二条第二項に規定する特定非営利活動法人が設置した治験審査委員会
四 医療関係者により構成された学術団体が設置した治験審査委員会
五 私立学校法(昭和二十四年法律第二百七十号)第三条に規定する学校法人(医療機関を有するものに限る。)が設置した治験審査委員会
六 独立行政法人通則法(平成十一年法律第百三号)第二条第一項に規定する独立行政法人(医療の提供等を主な業務とするものに限る。)が設置した治験審査委員会
七 国立大学法人法(平成十五年法律第百十二号)第二条第一項に規定する国立大学法人(医療機関を有するものに限る。)が設置した治験審査委員会
八 地方独立行政法人法(平成十五年法律第百十八号)第二条第一項に規定する地方独立行政法人(医療機関を有するものに限る。)が設置した治験審査委員会
2 前項第二号から第四号までに掲げる治験審査委員会は、その設置をする者(以下「治験審査委員会の設置者」という。)が次に掲げる要件を満たすものでなければならない。
一 定款その他これに準ずるものにおいて、治験審査委員会を設置する旨の定めがあること。
二 その役員(いかなる名称によるかを問わず、これと同等以上の職権又は支配力を有する者を含む。次号において同じ。)のうちに医師、歯科医師、薬剤師、看護師その他の医療関係者が含まれていること。
三 その役員に占める次に掲げる者の割合が、それぞれ三分の一以下であること。
イ 特定の医療機関の職員その他の当該医療機関と密接な関係を有する者
ロ 特定の法人の役員又は職員その他の当該法人と密接な関係を有する者
四 治験審査委員会の設置及び運営に関する業務を適確に遂行するに足りる財産的基礎を有していること。
五 財産目録、貸借対照表、損益計算書、事業報告書その他の財務に関する書類をその事務所に備えて置き、一般の閲覧に供していること。
六 その他治験審査委員会の業務の公正かつ適正な遂行を損なうおそれがないこと。
(治験審査委員会の構成等)
第二十八条 治験審査委員会は、次に掲げる要件を満たしていなければならない。
一 治験について倫理的及び科学的観点から十分に審議を行うことができること。
二 五名以上の委員からなること。
三 委員のうち、医学、歯学、薬学その他の医療又は臨床試験に関する専門的知識を有する者以外の者(次号及び第五号の規定により委員に加えられている者を除く。)が加えられていること。
四 委員のうち、実施医療機関と利害関係を有しない者が加えられていること。
五 委員のうち、治験審査委員会の設置者と利害関係を有しない者が加えられていること。
2 治験審査委員会の設置者は、次に掲げる事項について記載した手順書、委員名簿並びに会議の記録及びその概要を作成し、当該手順書に従って業務を行わせなければならない。
一 委員長の選任方法
二 会議の成立要件
三 会議の運営に関する事項
四 第三十一条第一項の適否の審査の実施時期に関する事項
五 会議の記録に関する事項
六 記録の保存に関する事項
七 その他必要な事項
3 治験審査委員会の設置者は、前項に規定する当該治験審査委員会の手順書、委員名簿及び会議の記録の概要を公表しなければならない。
4 治験審査委員会の設置者は、治験審査委員会の事務を行う者を選任しなければならない。
(治験審査委員会の会議)
第二十九条 次に掲げる委員は、審査の対象となる治験に係る審議及び採決に参加することができない。
一 治験依頼者の役員又は職員その他の治験依頼者と密接な関係を有する者
二 自ら治験を実施する者又は自ら治験を実施する者と密接な関係を有する者
三 実施医療機関の長、治験責任医師等又は治験協力者
2 審議に参加していない委員は、採決に参加することができない。
(治験審査委員会の審査)
第三十条 実施医療機関の長は、当該実施医療機関において治験を行うことの適否について、あらかじめ、第二十七条第一項の治験審査委員会の意見を聴かなければならない。
2 実施医療機関の長は、前項の治験審査委員会(当該実施医療機関の長が設置した第二十七条第一項第一号に掲げる治験審査委員会及び同項第五号から第八号までに掲げる治験審査委員会のうち当該実施医療機関を有する法人が設置したものを除く。)に調査審議を行わせることとする場合には、あらかじめ、次に掲げる事項を記載した文書により当該治験審査委員会の設置者との契約を締結しなければならない。
一 当該契約を締結した年月日
二 当該実施医療機関及び当該治験審査委員会の設置者の名称及び所在地
三 当該契約に係る業務の手順に関する事項
四 当該治験審査委員会が意見を述べるべき期限
五 被験者の秘密の保全に関する事項
六 その他必要な事項
3 前項の契約の締結については、第十二条第二項から第五項までの規定を準用する。この場合において、これらの規定中「治験の依頼をしようとする者」とあるのは「実施医療機関の長」と、「受託者」とあるのは「第二十七条第一項の治験審査委員会(当該実施医療機関の長が設置した同項第一号に掲げる治験審査委員会及び同項第五号から第八号までに掲げる治験審査委員会のうち当該実施医療機関を有する法人が設置したものを除く。)の設置者」と読み替えるものとする。
4 実施医療機関の長は、第一項の規定により第二十七条第一項の治験審査委員会の意見を聴くに当たり、治験を行うことの適否の判断の前提となる特定の専門的事項を調査審議させるため必要があると認めるときは、当該治験審査委員会の承諾を得て、当該専門的事項について当該治験審査委員会以外の治験審査委員会(第二十七条第一項各号に掲げるもの(同項第二号から第四号までに掲げるものにあっては、同条第二項各号に掲げる要件を満たすものに限る。)に限る。)の意見を聴くことができる。
5 実施医療機関の長は、前項の規定により意見を聴いた治験審査委員会(以下「専門治験審査委員会」という。)が意見を述べたときは、速やかに当該意見を第一項の規定により意見を聴いた治験審査委員会に報告しなければならない。
6 実施医療機関の長は、第四項の規定により専門治験審査委員会(当該実施医療機関の長が設置した第二十七条第一項第一号に掲げる治験審査委員会及び同項第五号から第八号までに掲げる治験審査委員会のうち当該実施医療機関を有する法人が設置したものを除く。)の意見を聴く場合には、あらかじめ、次に掲げる事項を記載した文書により当該専門治験審査委員会の設置者との契約を締結しなければならない。
一 当該契約を締結した年月日
二 当該実施医療機関及び当該専門治験審査委員会の設置者の名称及び所在地
三 当該契約に係る業務の手順に関する事項
四 当該専門治験審査委員会が調査審議を行う特定の専門的事項の範囲及び当該専門治験審査委員会が意見を述べるべき期限
五 被験者の秘密の保全に関する事項
六 その他必要な事項
7 前項の契約の締結については、第十二条第二項から第五項までの規定を準用する。この場合において、これらの規定中「治験の依頼をしようとする者」とあるのは「実施医療機関の長」と、「受託者」とあるのは「第三十条第五項に規定する専門治験審査委員会(当該実施医療機関の長が設置した第二十七条第一項第一号に掲げる治験審査委員会及び同項第五号から第八号までに掲げる治験審査委員会のうち当該実施医療機関を有する法人が設置したものを除く。)の設置者」と読み替えるものとする。
8 実施医療機関の長は、第一項又は第四項の規定により、第二十七条第一項の治験審査委員会(当該実施医療機関の長が設置した同項第一号に掲げる治験審査委員会を除く。)に意見を聴くときは、第二十八条第二項に規定する当該治験審査委員会の手順書及び委員名簿を入手しなければならない。
(継続審査等)
第三十一条 実施医療機関の長は、治験の期間が一年を越える場合には、一年に一回以上、当該実施医療機関において治験を継続して行うことの適否について、前条第一項の規定により意見を聴いた治験審査委員会(当該治験を継続して行うことの適否の判断の前提となる特定の専門的事項について前条第四項の規定により意見を聴いた専門治験審査委員会がある場合にあっては、同条第一項の規定により意見を聴いた治験審査委員会及び当該専門治験審査委員会)の意見を聴かなければならない。
2 実施医療機関の長は、第二十条第二項及び第三項、第二十六条の六第二項並びに第四十八条第二項及び第三項の規定により通知を受けたとき、第五十四条第三項の規定により報告を受けたときその他実施医療機関の長が必要があると認めたときは、当該実施医療機関において治験を継続して行うことの適否について、前条第一項の規定により意見を聴いた治験審査委員会(当該治験を継続して行うことの適否の判断の前提となる特定の専門的事項について前条第四項の規定により意見を聴いた専門治験審査委員会がある場合にあっては、同条第一項の規定により意見を聴いた治験審査委員会及び当該専門治験審査委員会)の意見を聴かなければならない。
3 前二項の規定により専門治験審査委員会の意見を聴く場合については、前条第五項の規定を準用する。
4 実施医療機関の長は、第二十六条の八第二項に規定するモニタリング報告書を受け取ったとき又は第二十六条の九第三項に規定する監査報告書を受け取ったときは、当該実施医療機関において治験が適切に行われているかどうか又は適切に行われたかどうかについて、前条第一項の規定により意見を聴いた治験審査委員会の意見を聴かなければならない。
(治験審査委員会の責務)
第三十二条 第二十七条第一項の治験審査委員会(以下この条において「治験審査委員会」という。)は、第三十条第一項の規定により実施医療機関の長から意見を聴かれたときは、審査の対象とされる治験が倫理的及び科学的に妥当であるかどうかその他当該治験が当該実施医療機関において行うのに適当であるかどうかを、次に掲げる資料に基づき審査し、文書により意見を述べなければならない。
一 第十条第一項各号又は第十五条の七各号に掲げる文書
二 被験者の募集の手順に関する資料
三 第七条第五項又は第十五条の四第四項に規定する情報その他治験を適正に行うために重要な情報を記載した文書
四 治験責任医師等となるべき者の履歴書
五 その他当該治験審査委員会が必要と認める資料
2 専門治験審査委員会は、第三十条第四項の規定により実施医療機関の長から意見を聴かれたときは、審査の対象とされる特定の専門的事項について前項各号に掲げる資料(当該専門治験審査委員会が必要と認めるものに限る。)に基づき審査し、文書により意見を述べなければならない。
3 治験審査委員会及び専門治験審査委員会は、前条第一項又は第二項の規定により実施医療機関の長から意見を聴かれたときは、治験審査委員会にあっては当該実施医療機関において当該治験が適切に行われているかどうかを調査した上で当該実施医療機関において治験を継続して行うことの適否を、専門治験審査委員会にあっては意見を聴かれた特定の専門的事項について調査した上で当該治験を継続して行うことの適否の判断の前提となる専門的事項をそれぞれ審査し、意見を聴かれた事項に係る事態の緊急性に応じて速やかに、文書により意見を述べなければならない。
4 治験審査委員会は、前条第四項の規定により、実施医療機関の長から意見を聴かれたときは、当該実施医療機関において当該治験が適切に行われているかどうか又は適切に行われていたかどうかについて審査し、文書により意見を述べなければならない。
5 第三十条第四項の規定により実施医療機関の長が専門治験審査委員会の意見を聴いた場合においては、治験審査委員会は、第一項又は第三項の規定により意見を述べるに当たり、同条第五項(前条第三項において準用する場合を含む。)の規定により報告された当該専門治験審査委員会の意見を踏まえて、これを行わなければならない。
6 実施医療機関の長は、第一項又は第三項の規定による治験審査委員会の意見を治験の依頼をしようとする者又は治験依頼者及び治験責任医師となるべき者又は治験責任医師に文書により通知しなければならない。
7 実施医療機関の長は、第一項、第三項又は第四項の規定による治験審査委員会の意見を自ら治験を実施しようとする者又は自ら治験を実施する者に文書により通知しなければならない。
8 第六項の規定による文書による通知については、第十条第二項から第五項までの規定を準用する。この場合において、これらの規定中「治験の依頼をしようとする者」とあるのは「実施医療機関の長」と、「実施医療機関の長」とあるのは「治験の依頼をしようとする者又は治験依頼者」と読み替えるものとする。
(治験審査委員会の意見)
第三十三条 実施医療機関は、第三十条第一項の規定により意見を聴いた治験審査委員会が、治験を行うことが適当でない旨の意見を述べたときは、治験の依頼を受け、又は治験の実施を承認してはならない。
2 実施医療機関は、第三十一条第一項又は第二項の規定により意見を聴いた治験審査委員会が、治験を継続して行うことが適当でない旨の意見を述べたときは、治験の契約を解除し、又は治験を中止しなければならない。
3 実施医療機関の長は、第三十一条第四項の規定により意見を聴いた治験審査委員会が、当該実施医療機関において当該治験が適切に行われていない旨又は適切に行われていなかった旨の意見を述べたときは、必要な措置を講じなければならない。
(記録の保存)
第三十四条 治験審査委員会を設置した者は、第二十八条第二項に規定する手順書、委員名簿並びに会議の記録及びその概要、第三十条第二項及び第六項の規定による契約に関する資料、第三十二条第一項各号に掲げる資料、同条第二項に規定する資料並びに第四十条第一項から第四項までの規定による治験審査委員会及び専門治験審査委員会に対する通知を、被験薬に係る医薬品についての製造販売の承認を受ける日(第二十四条第三項又は第二十六条の十第三項に規定する通知を受けたときは、通知を受けた日)又は治験の中止若しくは終了の後三年を経過した日のうちいずれか遅い日までの期間保存しなければならない。
第二節 実施医療機関
(実施医療機関の要件)
第三十五条 実施医療機関は、次に掲げる要件を満たしていなければならない。
一 十分な臨床観察及び試験検査を行う設備及び人員を有していること。
二 緊急時に被験者に対して必要な措置を講ずることができること。
三 治験責任医師等、薬剤師、看護師その他治験を適正かつ円滑に行うために必要な職員が十分に確保されていること。
(実施医療機関の長)
第三十六条 実施医療機関の長は、治験に係る業務に関する手順書を作成しなければならない。
2 実施医療機関の長は、当該実施医療機関における治験がこの省令、治験実施計画書、治験依頼者が治験を依頼する場合にあっては治験の契約書、自ら治験を実施する者が治験を実施する場合にあっては第十五条の七第五号から第十一号までに規定する文書及び前項の手順書に従って適正かつ円滑に行われるよう必要な措置を講じなければならない。
3 実施医療機関の長は、被験者の秘密の保全が担保されるよう必要な措置を講じなければならない。
(モニタリング等への協力)
第三十七条 実施医療機関の長は、治験依頼者が実施し、又は自ら治験を実施する者が実施させるモニタリング及び監査並びに第二十七条第一項の治験審査委員会及び第三十条第五項の専門治験審査委員会(専門治験審査委員会にあっては、第三十条第四項の規定により意見を聴く場合に限る。以下「治験審査委員会等」という。)による調査に協力しなければならない。
2 実施医療機関の長は、前項のモニタリング、監査又は調査が実施される際には、モニター、監査担当者又は治験審査委員会等の求めに応じ、第四十一条第二項各号に掲げる治験に関する記録を閲覧に供しなければならない。
(治験事務局)
第三十八条 実施医療機関の長は、治験に係る業務に関する事務を行う者を選任しなければならない。
(治験使用薬の管理)
第三十九条 治験薬管理者(治験薬を管理する者をいう。)は、第十六条第六項又は第二十六条の二第六項の手順書に従って治験使用薬を適切に管理しなければならない。
(業務の委託等)
第三十九条の二 実施医療機関(自ら治験を実施する者が治験を実施する場合にあっては、治験責任医師又は実施医療機関。以下この条において同じ。)は、治験の実施に係る業務の一部を委託する場合には、次に掲げる事項を記載した文書により当該業務を受託する者との契約を締結しなければならない。
一 当該委託に係る業務の範囲
二 当該委託に係る業務の手順に関する事項
三 前号の手順に基づき当該委託に係る業務が適正かつ円滑に行われているかどうかを実施医療機関が確認することができる旨
四 当該受託者に対する指示に関する事項
五 前号の指示を行った場合において当該措置が講じられたかどうかを実施医療機関が確認することができる旨
六 当該受託者が実施医療機関に対して行う報告に関する事項
七 その他当該委託に係る業務について必要な事項
(治験の中止等)
第四十条 実施医療機関の長は、第二十条第二項及び第三項の規定により治験依頼者から又は第二十六条の六第二項の規定により自ら治験を実施する者から通知を受けたときは、直ちにその旨を治験審査委員会等に文書により通知しなければならない。
2 実施医療機関の長は、第二十四条第二項の規定により治験依頼者から若しくは第二十六条の十第二項の規定により自ら治験を実施する者から治験を中断し、若しくは中止する旨の通知を受けたとき又は第二十四条第三項の規定により治験依頼者から申請書に添付しないことを決定した旨の通知若しくは第二十六条の十第三項の規定により自ら治験を実施する者から申請書に添付されないことを知った旨の通知を受けたときは、速やかにその旨及びその理由を治験責任医師及び治験審査委員会等に文書により通知しなければならない。
3 実施医療機関の長は、第四十九条第二項の規定により治験責任医師から治験を中断し、又は中止する旨の報告を受けた場合は、速やかにその旨及びその理由を治験審査委員会等及び治験依頼者に文書により通知しなければならない。
4 実施医療機関の長は、第四十九条第三項の規定により治験責任医師から治験を終了する旨の報告を受けたときは、その旨及びその結果の概要を治験審査委員会等及び治験依頼者に通知しなければならない。
5 第三項の規定による文書による通知については、第十条第二項から第五項までの規定を準用する。この場合において、これらの規定中「治験の依頼をしようとする者」とあるのは「実施医療機関の長」と、「実施医療機関の長」とあるのは「治験依頼者」と読み替えるものとする。
(記録の保存)
第四十一条 実施医療機関の長は、記録保存責任者を置かなければならない。
2 前項の記録保存責任者は、次に掲げる治験に関する記録(文書を含む。)を被験薬に係る医薬品についての製造販売の承認を受ける日(第二十四条第三項又は第二十六条の十第三項の規定により通知を受けたときは、通知を受けた日後三年を経過した日)又は治験の中止若しくは終了の後三年を経過した日のうちいずれか遅い日までの期間保存しなければならない。
一 原資料
二 契約書又は承認書、同意文書及び説明文書その他この省令の規定により実施医療機関に従事する者が作成した文書又はその写し
三 治験実施計画書、第三十二条第一項から第三項までの規定により治験審査委員会等から入手した文書その他この省令の規定により入手した文書
四 治験使用薬の管理その他の治験に係る業務の記録
第三節 治験責任医師
(治験責任医師の要件)
第四十二条 治験責任医師は、次に掲げる要件を満たしていなければならない。
一 治験を適正に行うことができる十分な教育及び訓練を受け、かつ、十分な臨床経験を有すること。
二 治験実施計画書、治験薬概要書及び第十六条第七項又は第二十六条の二第七項に規定する文書に記載されている治験使用薬の適切な使用方法に精通していること。
三 治験を行うのに必要な時間的余裕を有すること。
(治験分担医師等)
第四十三条 治験責任医師は、当該治験に係る治験分担医師又は治験協力者が存する場合には、分担する業務の一覧表を作成しなければならない。
2 治験責任医師は、治験分担医師及び治験協力者に治験の内容について十分に説明するとともに、第二十条第二項及び第三項の規定により通知された事項、第二十六条の六第二項の規定により通知した事項その他分担させる業務を適正かつ円滑に行うために必要な情報を提供しなければならない。
(被験者となるべき者の選定)
第四十四条 治験責任医師等は、次に掲げるところにより、被験者となるべき者を選定しなければならない。
一 倫理的及び科学的観点から、治験の目的に応じ、健康状態、症状、年齢、同意の能力等を十分に考慮すること。
二 同意の能力を欠く者にあっては、被験者とすることがやむを得ない場合を除き、選定しないこと。
三 治験に参加しないことにより不当な不利益を受けるおそれがある者を選定する場合にあっては、当該者の同意が自発的に行われるよう十分な配慮を行うこと。
(被験者に対する責務)
第四十五条 治験責任医師等は、治験使用薬の適正な使用方法を被験者に説明し、かつ、必要に応じ、被験者が治験使用薬を適正に使用しているかどうかを確認しなければならない。
2 治験責任医師等は、被験者が他の医師により治療を受けている場合には、被験者の同意の下に、被験者が治験に参加する旨を当該他の医師に通知しなければならない。
3 実施医療機関の長及び治験責任医師等は、被験者に生じた有害事象に対して適切な医療が提供されるよう、事前に、必要な措置を講じておかなければならない。
4 治験責任医師等は、被験者に有害事象が生じ、治療が必要であると認めるときは、その旨を被験者に通知しなければならない。
(治験実施計画書からの逸脱)
第四十六条 治験責任医師等は、治験審査委員会が事前に承認した治験実施計画書を遵守して、治験を実施しなければならない。
2 治験責任医師は、被験者の緊急の危険を回避するためその他医療上やむを得ない理由により治験実施計画書に従わなかった場合には、全てこれを記録し、その旨及びその理由を記載した文書を直ちに治験依頼者が治験を依頼する場合にあっては治験依頼者及び実施医療機関の長に、自ら治験を実施する者が治験を実施する場合にあっては実施医療機関の長に提出しなければならない。
3 治験依頼者が治験を依頼する場合における前項の規定による文書の提出については、第十条第二項から第五項までの規定を準用する。この場合において、これらの規定中「治験の依頼をしようとする者」とあるのは「治験責任医師」と、「実施医療機関の長」とあるのは「治験依頼者」と読み替えるものとする。
(症例報告書)
第四十七条 治験責任医師等は、治験実施計画書に従って正確に症例報告書を作成し、これに氏名を記載しなければならない。
2 治験責任医師等は、症例報告書の記載を変更し、又は修正するときは、これにその日付及び氏名を記載しなければならない。
3 治験責任医師は、治験分担医師が作成した症例報告書を点検し、内容を確認した上で、これに氏名を記載しなければならない。
(治験中の副作用等報告)
第四十八条 治験責任医師は、治験の実施状況の概要を、適宜、実施医療機関の長に文書により報告しなければならない。
2 治験依頼者が治験を依頼する場合にあっては、治験責任医師は、治験使用薬の副作用によると疑われる死亡その他の重篤な有害事象の発生を認めたときは、直ちに実施医療機関の長に報告するとともに、治験依頼者に通知しなければならない。この場合において、治験依頼者、実施医療機関の長又は治験審査委員会等から更に必要な情報の提供を求められたときは、当該治験責任医師はこれに応じなければならない。
3 自ら治験を実施する者が治験を実施する場合にあっては、治験責任医師は、治験使用薬の副作用によると疑われる死亡その他の重篤な有害事象の発生を認めたときは、直ちに実施医療機関の長(一つの実施計画書に基づき共同で複数の実施医療機関において治験を実施する場合には他の実施医療機関の治験責任医師を含む。)に報告するとともに、治験薬提供者に通知しなければならない。この場合において、治験薬提供者、実施医療機関の長又は治験審査委員会等から更に必要な情報の提供を求められたときは、当該治験責任医師はこれに応じなければならない。
(治験の中止等)
第四十九条 治験責任医師は、第四十条第二項の通知により治験が中断され、又は中止されたときは、被験者に速やかにその旨を通知するとともに、適切な医療の提供その他必要な措置を講じなければならない。
2 治験責任医師は、自ら治験を中断し、又は中止したときは、実施医療機関の長に速やかにその旨及びその理由を文書により報告しなければならない。
3 治験責任医師は、治験を終了したときは、実施医療機関の長にその旨及びその結果の概要を文書により報告しなければならない。
第四節 被験者の同意
(文書による説明と同意の取得)
第五十条 治験責任医師等は、被験者となるべき者を治験に参加させるときは、あらかじめ治験の内容その他の治験に関する事項について当該者の理解を得るよう、文書により適切な説明を行い、文書により同意を得なければならない。
2 被験者となるべき者が同意の能力を欠くこと等により同意を得ることが困難であるときは、前項の規定にかかわらず、被験者となるべき者の代諾者の同意を得ることにより、当該被験者となるべき者を治験に参加させることができる。
3 治験責任医師等は、前項の規定により被験者となるべき者の代諾者の同意を得た場合には、代諾者の同意に関する記録及び代諾者と被験者との関係についての記録を作成しなければならない。
4 治験責任医師等は、当該被験者に対して治験薬の効果を有しないと予測される治験においては、第二項の規定にかかわらず、同意を得ることが困難な被験者となるべき者を治験に参加させてはならない。ただし、第七条第二項又は第十五条の四第二項に規定する場合は、この限りではない。
5 治験責任医師等は、説明文書の内容その他治験に関する事項について、被験者となるべき者(被験者となるべき者の代諾者の同意を得る場合にあっては、当該者。次条から第五十三条までにおいて同じ。)に質問をする機会を与え、かつ、当該質問に十分に答えなければならない。
(説明文書)
第五十一条 治験責任医師等は、前条第一項の説明を行うときは、次に掲げる事項を記載した説明文書を交付しなければならない。
一 当該治験が試験を目的とするものである旨
二 治験の目的
三 治験責任医師の氏名及び連絡先
四 治験の方法
五 予測される治験薬による被験者の心身の健康に対する利益(当該利益が見込まれない場合はその旨)及び予測される被験者に対する不利益
六 他の治療方法に関する事項
七 治験に参加する期間
八 治験の参加をいつでも取りやめることができる旨
九 治験に参加しないこと又は参加を取りやめることにより被験者が不利益な取扱いを受けない旨
十 被験者の秘密が保全されることを条件に、モニター、監査担当者及び治験審査委員会等が原資料を閲覧できる旨
十一 被験者に係る秘密が保全される旨
十二 健康被害が発生した場合における実施医療機関の連絡先
十三 健康被害が発生した場合に必要な治療が行われる旨
十四 健康被害の補償に関する事項
十五 当該治験の適否等について調査審議を行う治験審査委員会の種類、各治験審査委員会において調査審議を行う事項その他当該治験に係る治験審査委員会に関する事項
十六 被験者が負担する治験の費用があるときは、当該費用に関する事項
十七 当該治験に係る必要な事項
2 説明文書には、被験者となるべき者に権利を放棄させる旨又はそれを疑わせる記載及び治験依頼者、自ら治験を実施する者、実施医療機関、治験責任医師等の責任を免除し、若しくは軽減させる旨又はそれを疑わせる記載をしてはならない。
3 説明文書には、できる限り平易な表現を用いなければならない。
(同意文書等への署名等)
第五十二条 第五十条第一項又は第二項に規定する同意は、被験者となるべき者が説明文書の内容を十分に理解した上で、当該内容の治験に参加することに同意する旨を記載した文書(以下「同意文書」という。)に、説明を行った治験責任医師等及び被験者となるべき者(第三項に規定する立会人が立ち会う場合にあっては、被験者となるべき者及び立会人。次条において同じ。)が日付を記載して、これに署名しなければ、効力を生じない。
2 第五十条第一項又は第二項に規定する同意は、治験責任医師等に強制され、又はその判断に不当な影響を及ぼされたものであってはならない。
3 説明文書を読むことができない被験者となるべき者(第五十条第二項に規定する被験者となるべき者を除く。)に対する同条第一項に規定する説明及び同意は、立会人を立ち会わせた上で、しなければならない。
4 前項の立会人は、治験責任医師等及び治験協力者であってはならない。
(同意文書の交付)
第五十三条 治験責任医師等は、治験責任医師等及び被験者となるべき者が署名した同意文書の写しを被験者(代諾者の同意を得た場合にあっては、当該者。次条において同じ。)に交付しなければならない。
(被験者の意思に影響を与える情報が得られた場合)
第五十四条 治験責任医師等は、治験に継続して参加するかどうかについて被験者の意思に影響を与えるものと認める情報を入手した場合には、直ちに当該情報を被験者に提供し、これを文書により記録するとともに、被験者が治験に継続して参加するかどうかを確認しなければならない。この場合においては、第五十条第五項及び第五十二条第二項の規定を準用する。
2 治験責任医師は、前項の場合において、説明文書を改訂する必要があると認めたときは、速やかに説明文書を改訂しなければならない。
3 治験責任医師は、前項の規定により説明文書を改訂したときは、その旨を実施医療機関の長に報告するとともに、治験の参加の継続について改めて被験者の同意を得なければならない。この場合においては、第五十一条から前条までの規定を準用する。
(緊急状況下における救命的治験)
第五十五条 治験責任医師等は、第七条第三項又は第十五条の四第三項に規定する治験においては、次の各号の全てに該当する場合に限り、被験者となるべき者及び代諾者となるべき者の同意を得ずに当該被験者となるべき者を治験に参加させることができる。
一 被験者となるべき者に緊急かつ明白な生命の危険が生じていること。
二 現在における治療方法では十分な効果が期待できないこと。
三 被験薬の使用により被験者となるべき者の生命の危険が回避できる可能性が十分にあると認められること。
四 予測される被験者に対する不利益が必要な最小限度のものであること。
五 代諾者となるべき者と直ちに連絡を取ることができないこと。
2 治験責任医師等は、前項に規定する場合には、速やかに被験者又は代諾者となるべき者に対して当該治験に関する事項について適切な説明を行い、当該治験への参加について文書により同意を得なければならない。
第五章 再審査等の資料の基準
(再審査等の資料の基準)
第五十六条 法第十四条又は第十九条の二の承認を受けた者が行う医薬品の臨床試験の実施に係る法第十四条第三項(法第十四条の二の二第五項において読み替えて適用する場合に限る。)、第十四条の四第五項及び第十四条の六第四項に規定する資料の収集及び作成については、第四条から第六条まで、第七条(第三項第一号を除く。)、第九条、第十条(第一項第二号を除く。)、第十一条から第十五条まで、第十六条から第二十三条まで、第二十四条第一項及び第二項、第二十五条、第二十六条並びに第二十七条から第五十五条までの規定を準用する。この場合において、これらの規定(見出しを含み、第十六条第二項ただし書を除く。)中「治験」とあるのは「製造販売後臨床試験」と、「治験実施計画書」とあるのは「製造販売後臨床試験実施計画書」と、「治験責任医師」とあるのは「製造販売後臨床試験責任医師」と、「治験国内管理人」とあるのは「製造販売後臨床試験国内管理人」と、「治験調整医師」とあるのは「製造販売後臨床試験調整医師」と、「治験調整委員会」とあるのは「製造販売後臨床試験調整委員会」と、「治験分担医師」とあるのは「製造販売後臨床試験分担医師」と、「治験責任医師等」とあるのは「製造販売後臨床試験責任医師等」と、「治験依頼者」とあるのは「製造販売後臨床試験依頼者」と、「治験薬管理者」とあるのは「製造販売後臨床試験薬管理者」と、「治験協力者」とあるのは「製造販売後臨床試験協力者」と、「治験審査委員会」とあるのは「製造販売後臨床試験審査委員会」と、「専門治験審査委員会」とあるのは「専門製造販売後臨床試験審査委員会」と、「治験審査委員会等」とあるのは「製造販売後臨床試験審査委員会等」と、「治験使用薬」とあるのは「製造販売後臨床試験使用薬」と、「治験使用薬等」とあるのは「製造販売後臨床試験使用薬等」と、これらの規定(見出しを含み、第十一条、第十六条の見出し及び同条第一項、第二項並びに第五項から第七項まで、第十七条(見出しを含む。)並びに第三十九条(見出しを含む。)の規定を除く。)中「治験薬」とあるのは「製造販売後臨床試験薬」と、第七条第一項第二号中「全部又は一部」とあるのは「一部」と、第十一条中「治験薬」とあるのは、「被験者、製造販売後臨床試験責任医師等又は製造販売後臨床試験協力者が被験薬及び対照薬の識別をできない状態(以下「盲検状態」という。)にした製造販売後臨床試験薬」と、第十二条第一項及び第十三条第一項中「全部又は一部」とあるのは「一部」と、第十六条の見出し及び同条第一項、第二項、第五項及び第七項中「治験薬」とあるのは「盲検状態にした製造販売後臨床試験薬」と、同条第一項第一号中「治験用」とあるのは「製造販売後臨床試験用」と、同条第二項ただし書中「被験者、治験責任医師等若しくは治験協力者が被験薬及び対照薬の識別をできない状態」とあるのは「盲検状態」と、「拡大治験」とあるのは「拡大製造販売後臨床試験」と、同条第二項第一号中「予定される」とあるのは「承認されている」と、第十七条(見出しを含む。)中「治験薬」とあるのは「盲検状態にした製造販売後臨床試験薬」と、第二十条第二項中「法第八十条の二第六項に規定する事項」とあるのは「法第六十八条の十第一項に規定する事項(医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律施行規則第二百二十八条の二十第一項第一号及び第二号に規定する事項であって当該製造販売後臨床試験において発生したものに限る。)」と、「当該被験薬について初めて治験の計画を届け出た日」とあるのは「当該被験薬に係る医薬品の製造販売の承認の際に厚生労働大臣が指定した日」と、同条第三項中「治験薬概要書」とあるのは「添付文書若しくは注意事項等情報」と、「直ちにその旨を治験責任医師」とあるのは「直ちにその旨を当該製造販売後臨床試験責任医師」と、同条第四項中「治験実施計画書及び治験薬概要書」とあるのは「製造販売後臨床試験実施計画書」と、第二十六条第一項中「に係る医薬品についての製造販売の承認(法第十四条の二の二第一項の規定により条件及び期限を付したものを除く。第二十六条の十二、第三十四条及び第四十一条第二項において同じ。)を受ける日(第二十四条第三項の規定により通知したときは、通知した日後三年を経過した日)又は治験の中止若しくは終了の後三年を経過した日のうちいずれか遅い日までの期間」とあるのは「の再審査又は再評価が終了した日後五年間」と、第三十四条中「に係る医薬品についての製造販売の承認を受ける日(第二十四条第三項又は第二十六条の十第三項に規定する通知を受けたときは、通知を受けた日)又は治験の中止若しくは終了の後三年を経過した日のうちいずれか遅い日までの期間」とあるのは「の再審査又は再評価が終了する日まで」と、第三十八条見出し中「治験事務局」とあるのは「製造販売後臨床試験事務局」と、第三十九条(見出しを含む。)中「治験薬」とあるのは「盲検状態にした製造販売後臨床試験薬」と、「第十六条第六項又は第二十六条の二第六項」とあるのは「第十六条第六項」と、第四十条第一項中「第二十条第二項及び第三項の規定により治験依頼者から又は第二十六条の六第二項の規定により自ら治験を実施する者」とあるのは「製造販売後臨床試験依頼者」と、同条第二項中「第二十四条第二項の規定により治験依頼者から若しくは第二十六条の十第二項の規定により自ら治験を実施する者」とあるのは「製造販売後臨床試験依頼者」と、「通知を受けたとき又は第二十四条第三項の規定により治験依頼者から申請書に添付しないことを決定した旨の通知若しくは第二十六条の十第三項の規定により自ら治験を実施する者から申請書に添付されないことを知った旨の通知」とあるのは「通知」と、第四十一条第二項中「に係る医薬品についての製造販売の承認を受ける日(第二十四条第三項又は第二十六条の十第三項の規定により通知を受けたときは、通知を受けた日後三年を経過した日)又は治験の中止若しくは終了の後三年を経過した日のうちいずれか遅い日までの期間」とあるのは「の再審査又は再評価が終了する日まで」と、第四十二条第二号中「治験実施計画書、治験薬概要書」とあるのは「製造販売後臨床試験実施計画書」と読み替えるものとする。
第六章 治験の依頼等の基準
(法第八十条の二第一項の厚生労働省令で定める基準)
第五十七条 法第八十条の二第一項の厚生労働省令で定める基準は、第四条第一項、第五条、第七条第一項(第九号及び第十一号から第十三号までを除く。)、第八条第一項、第十一条、第十三条(同条第一項第十号、第十二号か
ら第十五号まで及び第十七号を除く。)、第十四条及び第十五条の規定を準用する。この場合において、第四条第一項中「実施医療機関及び治験責任医師の選定、治験使用薬の管理、治験使用薬等の副作用情報等の収集、記録の保存その他の治験の依頼及び管理に係る」とあるのは「治験使用薬の管理及び記録の保存の」と、第五条中「試験その他治験の依頼をするために必要な試験」とあるのは「試験」と、第十三条第一項中「前条の規定により」とあるのは「治験の依頼及び管理に係る」と読み替えるものとする。
(法第八十条の二第四項の厚生労働省令で定める基準)
第五十八条 治験の依頼を受けた者に係る法第八十条の二第四項の厚生労働省令で定める基準は、第二十七条から第五十五条まで(第二十九条第一項第二号、第三十一条第四項、第三十二条第四項及び第七項、第三十三条第三項並びに第四十八条第三項を除く。)の規定を準用する。
2 自ら治験を実施する者に係る法第八十条の二第四項の厚生労働省令で定める基準は、第十五条の二第一項、第十五条の三、第十五条の四第一項(第九号及び第十一号から第十三号までを除く。)、第十五条の五第一項、第十五条の七(第九号、第十号及び第十二号から第十四号までを除く。)、第十五条の九、第二十六条の二(第一項第五号及び第七項を除く。)、第二十六条の七第一項及び第三項、第二十六条の十二(第一号から第四号までを除く。)、第二十七条から第五十五条まで(第二十九条第一項第一号、第三十二条第六項及び第八項並びに第四十八条第二項を除く。)の規定を準用する。この場合において、第十五条の二第一項中「治験実施計画書の作成、治験使用薬の管理、治験使用薬等の副作用情報等の収集、記録の保存その他の治験の実施の準備及び管理に係る」とあるのは「治験使用薬の管理及び記録の保存の」と、第十五条の三中「試験その他治験を実施するために必要な試験」とあるのは「試験」と、第二十六条の二第五項中「製造数量等の製造に関する」とあるのは「製造数量の」と、「安定性等の品質」とあるのは「品質」と、第二十六条の十二中「適切に保存」とあるのは「保存」と読み替えるものとする。
(法第八十条の二第五項の厚生労働省令で定める基準)
第五十九条 法第八十条の二第五項の厚生労働省令で定める基準は、第十六条(第一項第五号及び第七項を除く。)、第二十一条第一項並びに第二十六条第一項(第一号から第四号までを除く。)及び第二項の規定を準用する。この場合において、第十六条第五項中「製造数量等の製造に関する」とあるのは「製造数量の」と、「安定性等の品質」とあるのは「品質」と、第二十六条第一項中「適切に保存」とあるのは「保存」と読み替えるものとする。
附 則 抄
(施行期日)
第一条 この省令は、平成九年四月一日から施行する。
(承認審査資料の基準に関する経過措置)
第二条 法第十四条第三項に規定する資料のうち、この省令の施行前に収集され、又は作成されたもの及びこの省令の施行の際現に収集され、又は作成されているものについては、第三条中「次条から第五十五条までの規定の定めるところ」とあるのは「第三十条第一項、第三十五条、第四十四条、第四十七条第一項、第五十条第一項及び第二項の規定の定めるところ並びに薬事法施行規則等の一部を改正する省令(平成九年厚生省令第二十九号)第一条の規定による改正前の薬事法施行規則(昭和三十六年厚生省令第一号)第六十七条各号の規定の例」と、第五十条第一項中「文書により適切な説明を行い、文書により同意」とあるのは「適切な説明を行い、同意」とする。
2 法第十四条第三項に規定する資料のうち、平成九年六月三十日までに法第八十条の二第一項の治験の依頼が行われた治験又は同日までに同条第二項の規定により届け出られた計画に係る治験により収集され、又は作成されたもの(前項に規定するものを除く。)については、第三条中「次条」とあるのは「次条から第六条まで、第七条(第一項第九号を除く。)、第八条から第十二条まで、第十三条(第九号から第十三号まで及び第十五号を除く。)、第十四条、第十五条、第十六条(第六項を除く。)、第十七条から第二十条まで、第二十四条から第二十七条まで、第二十八条第二項及び第三項、第二十九条から第三十五条まで、第三十八条、第四十条から第五十条まで、第五十一条(第一項第十号を除く。)並びに第五十二条」とする。
3 法第十四条第三項に規定する資料のうち、平成十年三月三十一日までに法第八十条の二第一項の治験の依頼が行われた治験又は同日までに同条第二項の規定により届け出られた計画に係る治験により収集され、又は作成されたもの(第一項及び前項に規定するものを除く。)については、第三条中「次条」とあるのは「次条から第六条まで、第七条(第一項第九号を除く。)、第八条から第十二条まで、第十三条(第十二号及び第十五号を除く。)、第十四条から第二十条まで、第二十四条から第二十七条まで、第二十八条第二項及び第三項、第二十九条から第三十五条まで、第三十八条から第五十条まで、第五十一条(第一項第十号を除く。)並びに第五十二条」とする。
(再審査等の資料の基準に関する経過措置)
第三条 法第十四条の四第四項及び第十四条の五第四項に規定する資料のうち、平成九年六月三十日までに依頼が行われた市販後臨床試験により収集され、又は作成されたものについては、第五十六条中「第三項第一号」とあるのは「第一項第九号及び第三項第一号」と、「から第十六条まで」とあるのは「、第十二条、第十三条(第九号から第十三号まで及び第十五号を除く。)、第十四条、第十五条、第十六条(第六項を除く。)」と、「第二十三条」とあるのは「第二十条」と、「第二十五条」とあるのは「第二十五条から第二十七条まで、第二十八条第二項及び第三項、第二十九条から第三十五条まで、第三十八条、第四十条から第五十条まで、第五十一条(第一項第十号を除く。)並びに第五十二条」とする。
2 法第十四条の四第四項及び第十四条の五第四項に規定する資料のうち、平成十年三月三十一日までに依頼がなされた市販後臨床試験(前項に規定する市販後臨床試験を除く。)により収集され、又は作成されたものについては、第五十六条中「第三項第一号」とあるのは「第一項第九号及び第三項第一号」と、「第十一条」とあるのは「第十一条、第十二条、第十三条(第十二号及び第十五号を除く。)、第十四条」と、「第二十三条」とあるのは「第二十条」と、「第二十五条」とあるのは「第二十五条から第二十七条まで、第二十八条第二項及び第三項、第二十九条から第三十五条まで、第三十八条から第五十条まで、第五十一条(第一項第十号を除く。)並びに第五十二条」とする。
(法第八十条の二第一項の厚生省令で定める基準に関する経過措置)
第四条 この省令の施行前に治験の計画書であって第七条第一項(第二号から第四号まで及び第九号から第十三号までを除く。)の規定に適合するものが作成されていた場合における当該治験に係る法第八十条の二第一項に規定する治験の依頼については、第五十七条の規定にかかわらず、薬事法施行規則等の一部を改正する省令(平成九年厚生省令第二十九号)第一条の規定による改正前の薬事法施行規則(昭和三十六年厚生省令第一号。附則第六条において「旧施行規則」という。)第六十七条(第七号から第十一号までを除く。)の規定の例による。
2 平成九年四月一日から六月三十日までの間に法第八十条の二第二項の規定により届け出られた計画に係る治験(前項の場合における当該治験を除く。)に対する第五十七条の規定の適用については、第五十七条中「第十一号、第十三号」とあるのは、「第九号」とする。
3 平成九年七月一日から平成十年三月三十一日までの間に法第八十条の二第二項の規定により届け出られた計画に係る治験(第一項の場合における当該治験を除く。)に対する第五十七条の適用については、第五十七条中「第十一号、第十三号」とあるのは「第十一号」とする。
(法第八十条の二第四項の厚生省令で定める基準に関する経過措置)
第五条 この省令の施行前に治験の計画書であって第七条第一項(第二号から第四号まで及び第九号から第十三号までを除く。)の規定に適合するものが作成されていた場合における当該治験の依頼を受けた者に係る法第八十条の二第四項の治験をすることについては、第五十八条の規定にかかわらず、第三十条第一項、第三十五条、第四十四条、第四十七条第一項並びに第五十条第一項及び第二項の規定の例による。この場合において、第五十条第一項中「文書により適切な」とあるのは「適切な」とする。
2 平成九年四月一日から六月三十日までの間に法第八十条の二第一項の治験の依頼を受けた者又は同日までに同条第二項の規定により届け出られた計画に係る治験の依頼を受けた者(前項に規定する者を除く。)に対する第五十八条の適用については、第五十八条中「第二十七条」とあるのは「第二十七条、第二十八条第二項及び第三項、第二十九条から第三十五条まで、第三十八条、第四十条から第五十条まで、第五十一条(第一項第十号を除く。)並びに第五十二条」とする。
3 平成九年七月一日から平成十年三月三十一日までの間に法第八十条の二第一項の治験の依頼を受けた者(第一項及び前項に規定する治験の依頼を受けた者を除く。)に対する第五十八条の適用については、第五十八条中「第二十七条」とあるのは「第二十七条、第二十八条第二項及び第三項、第二十九条から第三十五条まで、第三十八条から第五十条まで、第五十一条(第一項第十号を除く。)並びに第五十二条」とする。
(法第八十条の二第五項の厚生省令で定める基準に関する経過措置)
第六条 この省令の施行前に治験の計画書であって第七条第一項(第二号から第四号まで及び第九号から第十三号までを除く。)の規定に適合するものが作成されていた場合における当該治験の依頼をした者に係る法第八十条の二第五項に規定する治験の管理については、第五十九条の規定にかかわらず、旧施行規則第六十七条第七号、第八号及び第十号の規定の例による。
2 平成九年四月一日から六月三十日までの間に法第八十条の二第一項の治験の依頼をした者又は同日までに同条第二項の規定により届け出られた計画に係る治験の依頼をした者(前項に規定する者を除く。)に対する第五十九条の適用については、第五十九条中「第七項」とあるのは「第六項及び第七項」と、「、第二十一条第一項並びに」とあるのは「並びに」とする。
3 平成九年七月一日から平成十年三月三十一日までの間に法第八十条の二第一項の治験の依頼をした者(第一項及び前項に規定する者を除く。)については、「、第二十一条第一項並びに」とあるのは「並びに」とする。
附 則 (平成一二年一〇月二〇日厚生省令第一二七号) 抄
(施行期日)
1 この省令は、内閣法の一部を改正する法律(平成十一年法律第八十八号)の施行の日(平成十三年一月六日)から施行する。
附 則 (平成一三年三月二六日厚生労働省令第三六号) 抄
(施行期日)
1 この省令は、書面の交付等に関する情報通信の技術の利用のための関係法律の整備に関する法律の施行の日(平成十三年四月一日)から施行する。
附 則 (平成一四年二月二二日厚生労働省令第一四号) 抄
1 この省令は、保健婦助産婦看護婦法の一部を改正する法律の施行の日(平成十四年三月一日)から施行する。
附 則 (平成一五年六月一二日厚生労働省令第一〇六号)
1 この省令は、薬事法及び採血及び供血あつせん業取締法の一部を改正する法律附則第一条第一号に掲げる規定の施行の日(平成十五年七月三十日)から施行する。
2 この省令の施行の際現に、この省令による改正前の医薬品の臨床試験の実施の基準に関する省令第十二条第一項及び第十三条第一項の規定に基づき締結された契約に基づき実施される治験に係る取扱いについては、なお従前の例による。
附 則 (平成一六年一二月二一日厚生労働省令第一七二号)
(施行期日)
第一条 この省令は、薬事法及び採血及び供血あつせん業取締法の一部を改正する法律第二条の規定の施行の日(平成十七年四月一日)から施行する。
(経過措置)
第二条 この省令の施行前に実施された又はこの省令の際現に実施されている医薬品の臨床試験については、この省令による改正後の医薬品の臨床試験の実施の基準に関する省令の規定にかかわらず、なお従前の例による。
附 則 (平成一八年三月三一日厚生労働省令第七二号) 抄
(施行期日)
第一条 この省令は、平成十八年四月一日から施行する。
(経過措置)
第二条 この省令の施行前に実施された又はこの省令の施行の際現に実施されている医薬品の臨床試験については、この省令による改正後の医薬品の臨床試験の実施の基準に関する省令(次条において「新令」という。)の規定にかかわらず、なお従前の例による。
第三条 この省令の施行前に治験実施計画書(医薬品の臨床試験の実施の基準に関する省令第七条第一項から第三項まで又は第十五条の四第一項から第三項までの規定に適合するものに限る。)又は製造販売後臨床試験実施計画書(この省令による改正前の医薬品の臨床試験の実施の基準に関する省令第五十六条において準用する第七条第一項から第三項まで(第三項第一号を除く。)の規定に適合するものに限る。)が作成された医薬品の臨床試験(前項に該当するものを除く。)については、新令の規定にかかわらず、なお従前の例による。
附 則 (平成二〇年二月二九日厚生労働省令第二四号) 抄
(施行期日)
第一条 この省令は、平成二十年四月一日から施行する。ただし、第二十条第二項及び第三項の改正規定、第二十八条第三項の改正規定、第三十一条第二項及び第四十条第一項の改正規定(「第二十条第二項」の下に「及び第三項」を加える部分に限る。)、第四十三条第二項の改正規定並びに第五十六条第一項の改正規定(「第二十条第二項」の下に「及び第三項」を加える部分に限る。)は、平成二十一年四月一日から施行する。
(経過措置)
第二条 この省令の施行前に実施された又はこの省令の施行の際現に実施されている医薬品の臨床試験については、この省令による改正後の医薬品の臨床試験の実施の基準に関する省令(次項において「新令」という。)の規定にかかわらず、なお従前の例による。
第三条 この省令の施行前に治験実施計画書(医薬品の臨床試験の実施の基準に関する省令第七条第一項から第三項まで又は第十五条の四第一項から第三項までの規定に適合するものに限る。)又は製造販売後臨床試験実施計画書(この省令による改正前の医薬品の臨床試験の実施の基準に関する省令第五十六条において準用する第七条第一項から第三項まで(第三項第一号を除く。)の規定に適合するものに限る。)が作成された医薬品の臨床試験(前条に該当するものを除く。)については、新令の規定にかかわらず、なお従前の例による。
附 則 (平成二〇年一一月二八日厚生労働省令第一六三号) 抄
(施行期日)
第一条 この省令は、一般社団法人及び一般財団法人に関する法律の施行の日(平成二十年十二月一日)から施行する。
附 則 (平成二一年三月三一日厚生労働省令第六八号) 抄
(施行期日)
第一条 この省令は、平成二十一年四月一日から施行する。
附 則 (平成二四年一二月二八日厚生労働省令第一六一号) 抄
(施行期日)
第一条 この省令は、公布の日から施行する。
(経過措置)
第三条 この省令の施行前に治験実施計画書が作成された治験についての治験依頼者に係る通知(基準省令第二十条第二項の通知をいう。以下同じ。)については、平成二十六年六月三十日までの間は、なお従前の例による。
2 前項の規定にかかわらず、同項の治験依頼者が平成二十六年六月三十日までの間に通知を行う場合において、当該通知については、当該治験依頼者の選択により、第二条の規定による改正後の基準省令(以下「新基準省令」という。)第二十条第二項の規定の適用を受けることができる。
3 新基準省令第二十条第二項の規定は、第一項の治験依頼者に係る通知については、平成二十六年七月一日から適用する。
4 新基準省令第二十条第二項の規定は、この省令の施行後に治験実施計画書が作成された治験についての治験依頼者に係る通知については、平成二十六年七月一日から適用する。
5 前項の治験依頼者に係る通知であって、平成二十六年六月三十日までの間に行われるものについては、第二条の規定による改正前の基準省令第二十条第二項の通知とみなして、同項の規定を適用する。
6 前項の規定にかかわらず、同項の通知については、同項の治験依頼者の選択により、新基準省令第二十条第二項の規定の適用を受けることができる。
附 則 (平成二六年七月三〇日厚生労働省令第八七号) 抄
(施行期日)
第一条 この省令は、薬事法等の一部を改正する法律(以下「改正法」という。)の施行の日(平成二十六年十一月二十五日)から施行する。
附 則 (平成二八年一月二二日厚生労働省令第九号)
この省令は、公布の日から施行する。
附 則 (平成二九年一〇月二六日厚生労働省令第一一六号) 抄
(施行期日)
第一条 この省令は、平成三十年四月一日から施行する。
附 則 (令和二年八月三一日厚生労働省令第一五五号) 抄
(施行期日)
第一条 この省令は、医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律等の一部を改正する法律(令和元年法律第六十三号)の施行の日(令和二年九月一日)から施行する。
附 則 (令和二年一二月二五日厚生労働省令第二〇八号) 抄
(施行期日)
第一条 この省令は、公布の日から施行する。
附 則 (令和三年一月二九日厚生労働省令第一五号) 抄
(施行期日)
第一条 この省令は、医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律等の一部を改正する法律(以下「改正法」という。)附則第一条第二号に規定する規定の施行の日(令和三年八月一日)から施行する。
附 則 (令和四年五月二〇日厚生労働省令第八四号) 抄
(施行期日)
1 この省令は、医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律等の一部を改正する法律(令和四年法律第四十七号)の公布の日から施行する。


転職サポート申し込みはこちら
- ステップ1
- まずは申し込み。入力は1分で終わります。
- ステップ2
- 希望にマッチした求人情報を提供します。
- ステップ3
- 書類選考・面接
- ステップ4
- 内定・入社
- 入社後もずっとサポート!

 年収査定はこちら
年収査定はこちら
- 転職を考える際、最も重要な条件の一つは給与です。
治験コーディネーター(CRC)への転職を考えている方々にとって、自身のキャリアや経験がどの程度評価されるのか、気になることでしょう。
こちらでは、あなたのプロフィールに基づき、治験コーディネーター(CRC)へ転職した場合の年収を予測します。

 合格予想はこちら
合格予想はこちら
- 「臨床経験が少ない」「転職回数が多い」といった理由で、選考に通過できるか不安になり、応募をためらう方も多いと思います。
こちらでは、あなたのプロフィールに基づき、治験コーディネーター(CRC)に応募した場合の書類選考の通過率や面接の合格率を予測します。

 掲示板で質問をする
掲示板で質問をする
- 些細な悩みや、ふとした疑問がある場合は、掲示板で気軽に質問しましょう。
面倒な登録は必要ありません。匿名で簡単に質問できます。多くの人の協力を得て、あなたの疑問を解決しましょう。
治験コーディネーター(CRC)や人事担当者などの専門家が回答いたします。









 CRC
CRC

 CRCの
CRCの
 CRCの
CRCの
 CRCの
CRCの
 CRCの
CRCの
 CRCに
CRCに
 CRCの
CRCの
 SMO
SMO
 SMO
SMO
 応募先の
応募先の
 治験
治験
 院内CRCと
院内CRCと



 2027年4月からの転職
2027年4月からの転職 CRC未経験特集
CRC未経験特集 CRC経験者特集
CRC経験者特集 看護師特集
看護師特集 臨床検査技師特集
臨床検査技師特集 保健師特集
保健師特集 薬剤師特集
薬剤師特集 管理栄養士特集
管理栄養士特集 臨床工学技士特集
臨床工学技士特集 理学療法士特集
理学療法士特集 作業療法士特集
作業療法士特集 臨床心理士特集
臨床心理士特集 MR特集
MR特集 CRA経験者特集
CRA経験者特集


 求人検索
求人検索  ログイン
ログイン 会員さま専用
会員さま専用 CRCの仕事
CRCの仕事  治験業界の研究
治験業界の研究 経験・資格別の注意点
経験・資格別の注意点 応募書類の作成
応募書類の作成 面接・適性検査の対策
面接・適性検査の対策 みんなのクチコミ
みんなのクチコミ みんなの質問と回答
みんなの質問と回答 転職成功事例
転職成功事例 マンガで分かるCRC
マンガで分かるCRC CRCばんくチャンネル
CRCばんくチャンネル 便利な機能
便利な機能 相談/年収査定/合格予想
相談/年収査定/合格予想 2027年から働くには?
2027年から働くには? 退職手続き
退職手続き 開催中のキャンペーン
開催中のキャンペーン 《CRCばんく》とは
《CRCばんく》とは